Chromosome Segregation in Higher Eukaryotes

- Mary Dasso, PhD, Head, Section on Cell Cycle Regulation
- Alexei Arnaoutov, PhD, Staff Scientist
- Vasilisa Aksenova, PhD, Visiting Fellow
- Sarine Markossian, PhD, Visiting Fellow
- Maia Ouspenskaia, DVM, Biologist
- Shane Chen, PhD, Postdoctoral Intramural Research Training Award Fellow
- Saroj Regmi, PhD, Postdoctoral Intramural Research Training Award Fellow
- Joseph Bareille, BA, Predoctoral Visiting Fellow
- Ka Chun Yau, BS, Predoctoral Visiting Fellow
- Carlos Echeverria, BS, Postbaccalaureate Intramural Research Training Award Fellow
- Alexandra Smith, BA, Postbaccalaureate Intramural Research Training Award Fellow
- Elizabeth Turcotte, BA, BS, Postbaccalaureate Intramural Research Training Award Fellow
We are interested in mechanisms of chromosome segregation, defects in which lead to aneuploidy, that is, an abnormal number of chromosomes. Several common birth defects, such as Down's syndrome, result from aneuploidy arising during meiotic cell divisions. Moreover, aneuploidy arising from mitotic divisions is a hallmark of many types of solid tumors. During interphase, chromosomes are enclosed within nuclei, and exchange of all molecules between this compartment and the rest of the cell occurs through nuclear pore complexes (NPCs). Surprisingly, NPC proteins and proteins involved in trafficking of molecules into and out of the nucleus have important roles in chromosome segregation; we are investigating these roles at a molecular level. Our studies have concentrated on a GTPase called Ran and on a family of small ubiquitin-like modifiers (SUMOs), which are indispensable for mitotic chromosome segregation.
We also recently reported that the IRBIT protein is an inhibitor of ribonucleotide reductase; IRBIT works through a novel mechanism and is vital for genomic integrity.
The ultimate goals of our studies are to understand how these pathways enable accurate chromosome segregation and to discover how they are coordinated with each other and with other aspects of cell physiology.
Mitotic roles of nuclear pore complex proteins
NPCs consist of about thirty distinct proteins called nucleoporins. Kinetochores are proteinaceous structures that assemble at the centromere of each sister chromatid during mitosis and serve as sites of spindle microtubule attachment. The relationship between NPCs and mitotic kinetochores is surprisingly intimate but poorly understood. During interphase, several kinetochore proteins stably bind to NPCs (e.g., Mad1, Mad2, Mps1). During mitosis, metazoan NPCs disassemble, and at least a third of nucleoporins, including the RanBP2 complex and the Nup107-160 complex, associate with kinetochores. We showed that the complexes play important roles in kinetochore function. Additional nucleoporins that do not associate with kinetochores have also been shown to have important mitotic roles, including Nup214, Nup98, and TPR.
Click image to enlarge.
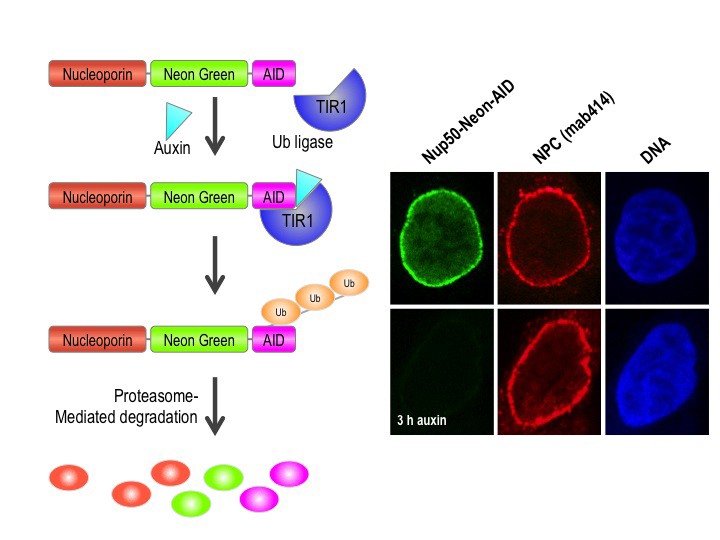
Figure 1. Auxin-induced degradation of AID–tagged nucleoporins
Cells expressing the TIR1 protein recognize proteins tagged with auxin-induced degron (AID) domains upon the addition of the plant hormone auxin. This leads to their rapid ubiquitination and destruction. We took advantage of this system by homozygously targeting endogenous nucleoporin genes with the AID tag and a fluorescent marker (neon green) in TIR1–expressing DLD1 cells. As shown on the right for the nucleoporin Nup50, we observe rapid and uniform degradation after auxin addition (left panels). Note that the nuclear pore is not generally disrupted, as indicated by staining with an antibody that recognizes a family of nucleoporins (mab414, middle panels).
The dual role of nucleoporins in controlling nuclear transport in interphase and spindle assembly in mitosis makes it difficult to precisely study their mitotic role. Nucleoporin depletion by RNAi knockdown both disrupts nuclear transport and, in many cases, causes cells to arrest in mitosis with disrupted kinetochore structures. Given that nucleoporins are long-lived proteins, a fast-reacting protein degradation system that depletes proteins at a post-translational level would be a preferable method to study their activities at discrete points during the cell cycle. To address this problem, we are currently constructing and testing auxin-induced degron (AID)–tagged lines for each of the human nucleoporin proteins in human cells that stably express the Transport Inhibitor Response 1 (TIR1) protein. Auxin family hormones bind to TIR and promote its interaction with the AID domain. TIR acts as a substrate-recognition subunit for ubiquitination by the ubiquitin ligase SCF, resulting in rapid (within 30 min) auxin-dependent proteasomal degradation of AID–tagged substrates in a manner that is reversible and tunable (Figure 1). By eliciting programmed degradation of nucleoporins at different cell-cycle stages, their functions in interphase and mitosis can be differentiated and characterized.
We are validating each of the AID–tagged lines to ensure they are homozygously tagged for each nucleoporin and that the tag moiety does not disrupt function in the absence of auxin. These tests include assays of cell proliferation, chromosome segregation, and nuclear trafficking (Figure 2). After validation, we plan to analyze the roles of individual nucleoporins and of nucleoporin subcomplexes in kinetochore function, mitotic progression, and spindle assembly.
Click image to enlarge.
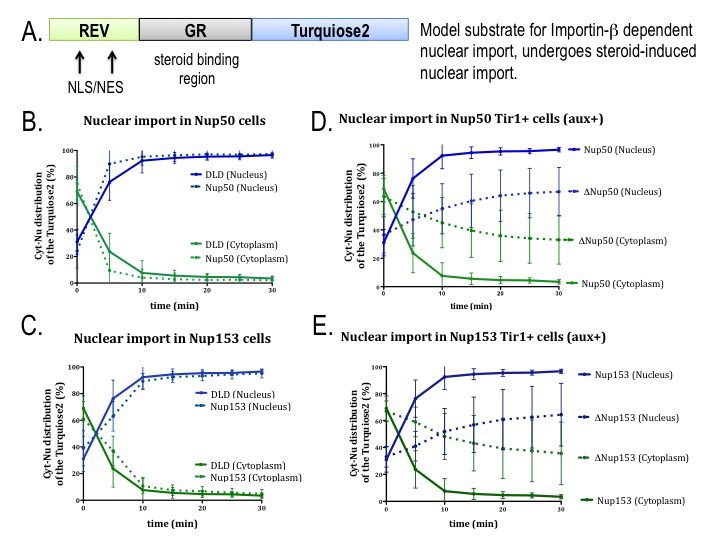
Figure 2. AID tagging of nucleoporins does not disrupt nuclear trafficking in the absence of auxin.
In order to validate the study of tagged nucleoporins, we assayed the capacity of cells expressing these proteins to import a model substrate that is imported to the nucleus by the transport receptor importin beta (A). Cell lines with tagged nucleoporins Nup50 (B) or Nup153 (C) show import rates similar to DLD1 parental cell lines. For both nucleoporins (D, E), import rates are slower after auxin addition (dotted lines) than in the absence of auxin (solid lines).
Mitotic regulation of the Ran GTPase
Ran is a Ras–family GTPase that plays critical roles in many cellular processes, including nucleo-cytoplasmic transport, nuclear envelope assembly, and mitotic spindle assembly. Ran alternates between GDP– and GTP–bound forms. In interphase cells, GTP–bound Ran (Ran-GTP) is the major form in nucleus while GDP–bound Ran (Ran-GDP) is the predominant form in cytoplasm. The asymmetrical distribution of Ran-GTP and Ran-GDP drives cargo transport between the nucleus and cytoplasm through karyopherins, a family of nuclear transport carrier proteins that bind to Ran-GTP. In mitosis, after nuclear envelope breakdown, Ran-GTP is concentrated in the region close to mitotic chromatin, while Ran-GDP is the major form distal to chromatin. The Ran-GTP gradient guides mitotic spindle assembly by releasing spindle assembly factors (SAFs) from karyopherins based on local Ran-GTP concentrations. In cells, the conversion of Ran-GDP to Ran-GTP is catalyzed by a Ran–specific guanine exchange factor (RanGEF) called RCC1 (Regulator of chromosome condensation 1) in vertebrates. The capacity of RCC1 to bind to chromatin establishes the asymmetrical distribution of Ran-GTP in interphase as well as the chromatin-centered Ran-GTP gradient in mitosis. Interestingly, RCC1’s association with chromatin is not static during the cell cycle and is regulated in a particularly dramatic fashion during anaphase in vertebrate systems. The regulation has not been correlated with post-translational modifications of RCC1, and the underlying molecular mechanism has not been reported.
RanBP1 is a highly conserved Ran-GTP–binding protein that acts as co-activator of RanGAP1 and can form a heterotrimeric complex with Ran and RCC1 in vitro. We found that RCC1 not associated with chromosomes during mitosis is sequestered and inhibited in RCC1/Ran/RanBP1 heterotrimeric complexes and that the sequestration is crucial for normal mitotic spindle assembly. In addition, RanBP1 complex formation competes with chromatin binding to regulate the distribution of RCC1 between the chromatin-associated and soluble fractions. Moreover, we identified a cell cycle–dependent phosphorylation on RanBP1 that modulates RCC1/Ran/RanBP1 heterotrimeric complex assembly and releases RCC1 to bind to chromatin; the phosphorylation is directly responsible for controlling RCC1 dynamics during anaphase. Together, our findings demonstrate novel roles of RanBP1 in spindle assembly and RCC1 regulation in mitosis. We are currently extending these findings to analyze whether RanBP1 plays an analogous role in mammalian cells during mitosis.
SUMO–family small ubiquitin-like modifiers in higher eukaryotes
SUMOs are ubiquitin-like proteins (Ubls) that become conjugated to substrates through a pathway that is biochemically similar to ubiquitination (Figure 3). SUMOylation is involved in many cellular processes, including DNA metabolism, gene expression, and cell-cycle progression. Vertebrate cells express three major SUMO paralogs (SUMO-1–3): mature SUMO-2 and SUMO-3 are 95% identical, while SUMO-1 is 45% identical to SUMO-2 or SUMO-3 (where they are functionally indistinguishable, we collectively call SUMO-2 and SUMO-3 SUMO-2/3). Like ubiquitin, SUMO-2/3 can be assembled into polymeric chains through the sequential conjugation of SUMOs to each other. Many SUMOylation substrates have been identified. SUMOylation promotes a variety of fates for individual targets, dependent upon the protein itself, the conjugated paralog, and whether the conjugated species contains a single SUMO or SUMO chains.
SUMOylation is dynamic owing to rapid turnover of conjugated species by SUMO proteases. Both post-translational processing of SUMO polypeptides and deSUMOylation are mediated by the same family of proteases, which play a pivotal role in determining the spectrum of SUMOylated species. This group of proteases is called Ubl–specific proteases (Ulp) in yeast and Sentrin-specific proteases (SENP) in vertebrates. There are two yeast Ulps (Ulp1p and Ulp2p/Smt4p) and six mammalian SENPs (SENP1, SENP2, SENP3, SENP5, SENP6, and SENP7). SENP1, SENP2, SENP3, and SENP5 form a Ulp1p–related sub-family, while SENP6 and SENP7 are more closely related to Ulp2p. Yeast Ulps have important roles in mitotic progression and chromosome segregation. We defined the enzymatic specificity of the vertebrate SENP proteins and analyzed their key biological roles.
Ulp1p localizes to NPCs, is encoded by an essential gene, and is important for SUMO processing, nucleocytoplasmic trafficking, and late steps in the ribosome biogenesis pathway. Humans possess two NPC–associated SENPs: SENP1 and SENP2. While SENP2 is dispensable for cell division, mammalian SENP1 was recently shown to play an essential role in mitotic progression. We are currently analyzing AID–tagged alleles of both SENP1 and SENP2 to assess their roles in both interphase (nuclear trafficking and gene expression) and during mitosis (kinetochore function and mitotic progression), as well as their dependence upon individual nucleoporins for their targeting to the interphase NPC.
Click image to enlarge.
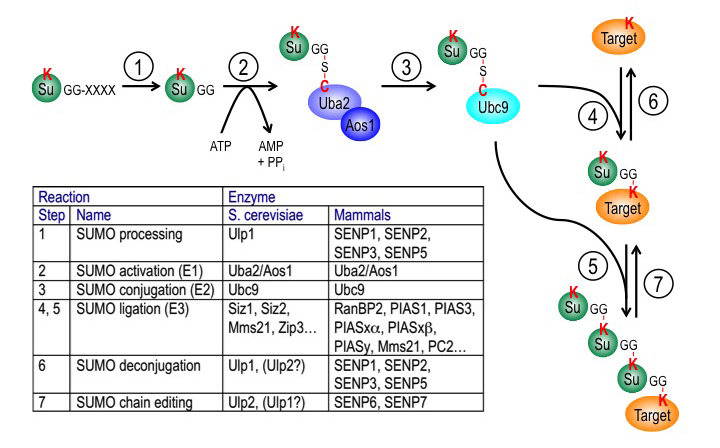
Figure 3. The SUMO pathway
SUMO proteins are post-translationally processed (step 1). Processed SUMO polypeptides possess a C-terminal diglycine motif, which is activated to form an ATP–dependent thioester linkage with the SUMO E1 enzyme, the Aos1/Uba2 heterodimer (step 2). The activated SUMO is transferred to thioester linkage on a conserved cysteine of the SUMO E2 enzyme Ubc9 (step 3). Finally, the activated SUMO becomes covalently linked through an isopeptide bond to lysine residues within cellular target proteins, a reaction that is typically promoted by SUMO ligases (E3 enzymes) acting in conjunction with Ubc9 (step 4). For some substrates, additional SUMOs can be added to form SUMO chains that can act as a signal for proteolytic degradation (step 5). Both mono-SUMOylation (step 6) and poly-SUMOylation (step 7) can be reversed by a family of SUMO–specific proteases that are also major catalysts of post-translational SUMO processing, called Ulps (Ubiquitin-like protein proteases) in yeast and SENPs (Sentrin-specific protease) in vertebrates. The inserted table provides the names of proteins involved in each of these steps in budding yeast and human cells. Note that many of these enzymes (Ubc9, RanBP2, Ulp1, SENP1, SENP2) associate to the nuclear pore complex.
Phosphorylation of Xenopus p31comet by IKK-beta potentiates mitotic checkpoint exit.
Given that it prevents chromosome mis-segregation and averts genomic instability, the spindle assembly checkpoint (SAC) is among the most important cellular surveillance pathways. To do this, the SAC inhibits premature separation of mitotic sister chromatids by monitoring interactions between kinetochores (KTs) and spindle microtubules (MTs). SAC components are essential in vertebrate cells because organisms that undergo open mitosis must re-establish KT-MT attachments after nuclear envelope breakdown (NEBD) in each cell division. During the process, unattached KTs transiently invoke the SAC during the interval between NEBD and MT attachment, so that subsequent SAC silencing plays a key role in determining the timing of anaphase onset. While SAC silencing is thus critical for mitotic progression in metazoan cells, it remains poorly understood. Found in higher eukaryotes, the p31comet protein plays an important role in SAC silencing. We used Xenopus egg extracts (XEEs) to investigate the mitotic roles and regulation of p31comet. Our observations suggest that endogenous p31comet is important for anaphase timing in this system and is regulated through mitotic phosphorylation. While several well established mitotic kinases did not efficiently modify p31comet in vitro, IKK-beta (Inhibitor of nuclear factor k-B kinase-beta) was an effective p31comet kinase. Depletion or inhibition of IKK-beta delayed mitotic exit of XEEs, and a phosphomimetic p31comet mutant showed increased activity in SAC silencing. Together, our experiments suggest that p31comet contributes to the timing of anaphase onset in XEE through antagonism of the SAC and that IKK-beta modifies p31comet to enhance its activity. Several previous reports established that IKK-beta plays a clear but under-appreciated role of within mitosis, a role that is distinct from its function in cellular stress and inflammatory signaling pathways. Our findings are among the first mechanistic insights into the nature of this role (Reference 1).
Development of novel assays for the quantitative assessment of chromosome instability
Most solid tumors are aneuploid, that is, they carry an abnormal number of chromosomes, and they missegregate whole chromosomes in a phenomenon termed chromosome instability (CIN). CIN is associated with poor prognosis in many cancer types, and targeting of CIN is an attractive strategy for anti-cancer therapeutics. The mechanisms causing CIN and its contributions to tumor initiation and growth are not well defined, partly because there is no straightforward, quantitative assays for CIN in human cells. To address this problem, we developed the first Human Artificial Chromosome (HAC)–based quantitative live-cell assay for mitotic chromosome segregation in mammalian cells, with which we can easily score the rates of CIN within one cell division under different experimental conditions (Reference 3). We constructed a HAC encoding copies of enhanced green fluorescent protein (eGFP) fused to the destruction box (DB) of hSecurin, a substrate of the anaphase-promoting complex/cyclosome (APC/C) ubiquitin ligase, which becomes active during anaphase to catalyze the proteolysis of critical mitotic target proteins. This HAC also contains tetracycline operator (tetO) arrays and sequences encoding the tetracycline repressor fused to monomeric cherry fluorescent protein (tetR-mCherry). We produced human U2OS cells (U2OS-Phoenix) carrying this HAC, in which we monitor HAC segregation in two ways. First, APC/C degrades the DB-eGFP fusion expressed from the HAC at anaphase onset, and DB-eGFP re-accumulates in the daughter cells after G1 phase, when APC/C becomes inactive. Daughter cells that do not obtain a copy of the HAC will thus be GFP–negative in the subsequent interphase (Figure 4). Second, because tetR-mCherry binds to the tetO arrays, the HAC itself could be followed by live imaging. Following the HAC by live-cell imaging experiments, we showed that U2OS-Phoenix cells have low inherent levels of CIN, but that HAC mis-segregation is markedly increased by treatment with Reversine, an inhibitor of the mitotic monopolar spindle 1 kinase (Mps1), and the microtubule agents Nocodazole and Taxol. In summary, we developed new assays to score CIN levels in human cells and showed that CIN levels increase upon chemical disruption of mitotic progression, demonstrating the utility of this assay for chemical screens of CIN–inducing compounds. The development of additional assays for CIN is ongoing, as are efforts to use the assays in high-throughput screens for genes and chemical compounds that can modulate CIN frequency.
Click image to enlarge.
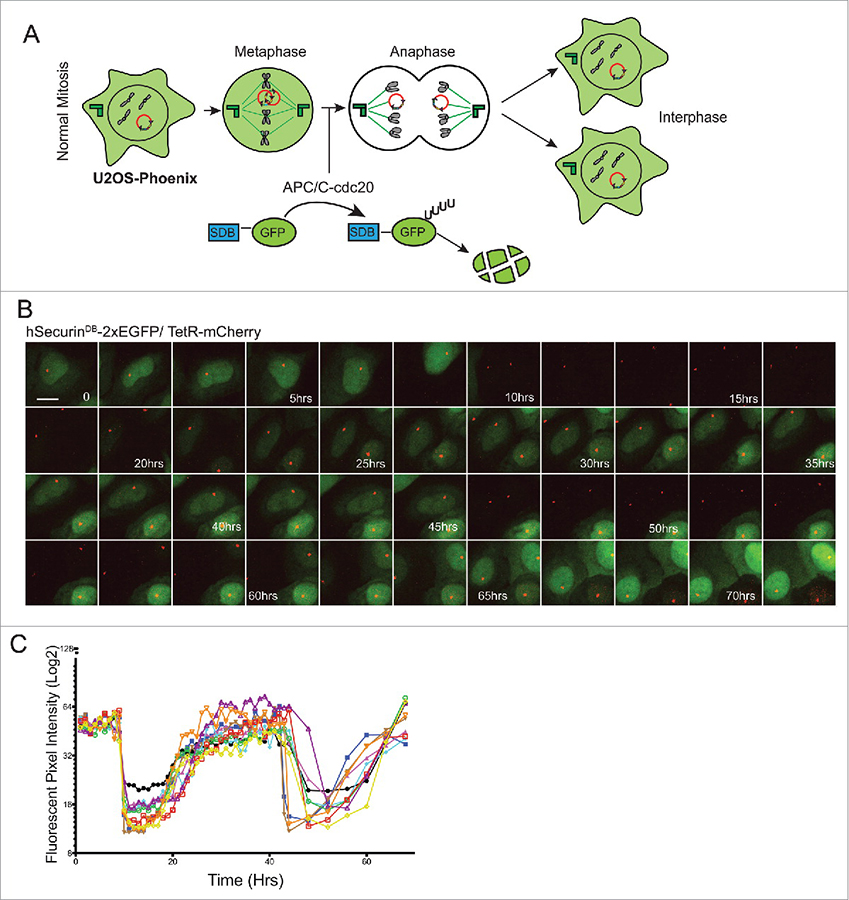
Figure 4. GFP expression in U2OS-Phoenix cells is coupled to the cell cycle.
A) Cartoon depicting U2OS-Phoenix cells undergoing one round of error-free mitosis. At the onset of anaphase, active APC/C-cdc20 recognizes and ubiquitinates DB–containing proteins, promoting their degradation. Thus, hSecurinDB-2xeGFP fusions expressed from HACs are rapidly degraded at the onset of anaphase, and re-accumulate in the two daughter cells after G1 phase. B) Still images from a live cell–imaging experiment following hSecurinDB-2xGFP and TetR-mCherry signals in U2OS-Phoenix cells, using a spinning disc confocal microscope. The images follow one cell in two rounds of consecutive mitosis. The GFP levels in the imaged cell and its daughter are coupled to the cell cycle. Size bars = 30 μm. C) The mean fluorescent intensity of the GFP signal was quantified in 10 cells from movies such as in (B) and plotted against time.
Analysis of IRBIT function in Drosophila
We discovered that the vertebrate IRBIT protein inhibits the catalytic activity of ribonucleotide reductase (RNR), the enzyme that catalyzes the production of deoxynucleotides for DNA synthesis. Similar biochemical relationships are conserved between mammalian and Drosophila melanogaster homologs of both proteins, allowing us to employ flies as a genetic system to analyze IRBIT biology. We generated flies lacking Drosophila IRBIT (dIRBIT–/– flies), which superficially appeared normal and were viable but sickly. In collaboration with the laboratories of Mihaela Serpe and Brian Oliver, we are particularly analyzing the role of IRBIT in tissue homeostasis within the fly midgut.
Additional Funding
- NICHD DIR Director’s Investigator Award
Publications
- Mo M, Arnaoutov A, Dasso M. Phosphorylation of Xenopus p31comet potentiates mitotic checkpoint exit. Cell Cycle 2015;14:3978-3985.
- Ryu H, Yoshida MM, Sridharan V, Kumagai A, Dunphy WG, Dasso M, Azuma Y. SUMOylation of the C-terminal domain of DNA topoisomerase IIa regulates the centromeric localization of Claspin. Cell Cycle 2015;14:2777-2784.
- Markossian S, Arnaoutov A, Saba NS, Larionov V, Dasso M. Quantitative assessment of chromosome instability induced through chemical disruption of mitotic progression. Cell Cycle 2016;15:1706-1714.
- Dasso M, Fontoura BM. Gating immunity and death at the nuclear pore complex. Cell 2016;166:1364-1366.
- Dasso M. Kar9 controls the cytoplasm by visiting the nucleus. Dev Cell 2016;36:360-361.
Collaborators
- Yoshiaki Azuma, PhD, University of Kansas, Lawrence, KS
- Vladimir Larionov, PhD, Developmental Therapeutics Branch, Center for Cancer Research, NCI, Bethesda, MD
- Brian C. Oliver, PhD, Laboratory of Cellular and Developmental Biology, NIDDK, Bethesda, MD
- Mihaela Serpe, PhD, Unit on Cellular Communication, NICHD, Bethesda, MD
Contact
For more information, email mdasso@helix.nih.gov or visit http://sccr.nichd.nih.gov.