Structural and Chemical Biology of Membrane Proteins

- Anirban Banerjee, PhD, Head, Unit on Structural and Chemical Biology of Membrane Proteins
- Eric Christenson, PhD, Postdoctoral Fellow (Contractor)
- Elliot Murphy, PhD, Postdoctoral Intramural Research Training Award Fellow
- Robbins Puthenveetil, PhD, Postdoctoral Intramural Research Training Award Fellow
- Elvis Ongey Legala, PhD, Visiting Fellow
- Kevin Choi, Postbaccalaureate Fellow
- Liam Healy, Postbaccalaureate Fellow
Molecular mechanism of post-translational protein lipidation by zDHHC protein S-acyltransferases
Post-translational modifications greatly expand the structural, chemical, and functional diversity of the proteome. Of these, protein lipidation, which collectively refers to covalent modification of proteins by lipids, constitutes a centrally important class of post-translational modification. Protein S-acylation, commonly known as protein palmitoylation, is a specific form of protein lipidation whereby long-chain fatty acids, typically C16, become covalently attached to cytosol-facing cysteines through a thioester linkage. Palmitoylation is one of the most pervasive and physiologically important post-translational modifications, and the targets of palmitoylation span a very wide range of proteins, ranging from ion channels to cell-surface receptors, neuronal scaffolding proteins, and small GTPases. The repertoire of palmitoylated proteins has expanded rapidly in recent years, with thousands of proteins now known to be part of the cellular 'palmitoylome.' The physico-chemical effect of palmitoylation is to alter the local hydrophobicity of the substrate protein. The thioester bond makes S-acylation unique in that it is a labile moiety and can be cleaved, in the cellular context, by thioesterase enzymes, which makes S-acylation one of the few dynamic post-translational modifications and unique among different forms of protein lipidation. The physiological effects of S-acylation are diverse and have critical cellular importance; for example, Ras, a small GTPase that is critical for cellular growth and differentiation and is mutated in about one-third of all human cancers, is palmitoylated at the Golgi and subsequently targeted to the plasma membrane by vesicular transport. Palmitoylated Ras localizes to cholesterol-rich domains on the plasma membrane. However, it is subsequently depalmitoylated by the thioesterase APT1, dissociates from the plasma membrane, and redistributes on endomembranes, including the Golgi. Such dynamic recycling of Ras is critical for its function. On the other hand, in recent work (see below), we showed that the Spike protein of SARS-CoV-2, the causative agent of COVID-19, is S-acylated, which is important in the viral life cycle.
Protein S-acylation is catalyzed by a large group of enzymes known as zDHHC palmitoyl acyltransferases (also referred to as DHHC enzymes or DHHC-PAT), so named because they contain a signature D-H-H-C motif (aspartate-histidine-histidine-cysteine) in a cysteine-rich domain (CRD) in an intracellular loop (Figure 1). These are low-abundance polytopic integral membrane proteins localized to a variety of cellular compartments. Humans have 23 DHHC-PATs encoded in their genome. Beyond the shared DHHC domain, DHHC-PATs vary considerably; some possess ankyrin repeats (structural protein motifs that mediate protein-protein interactions), a few have six transmembrane helices instead of the usual four, and at least one forms a functional heterodimer with a cytoplasmic auxiliary subunit. To date, no consensus sequences have been reported for palmitoylation. A specific DHHC-PAT can palmitoylate many substrates, and, conversely, a given substrate can be palmitoylated by many DHHC-PATs. Such redundancy has been one of the most intriguing aspects of DHHC-PATs and makes it difficult to assign substrates by overexpression/knockout strategies, given that, in the absence of one DHHC-PAT enzyme, others can take over. However, this does not necessarily reflect the true enzyme-substrate relationship. The situation is even more confounded by the lack of specific inhibitors of DHHC-PATs. Even though 2-bromopalmitate is widely used as a global inhibitor of DHHC-PATs, it has been shown that it also broadly targets other proteins involved in lipid metabolism.
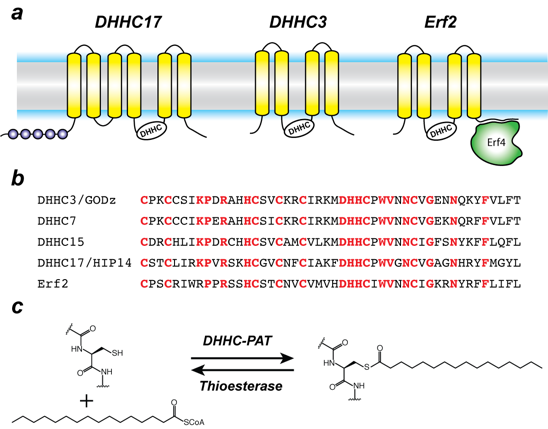
Figure 1. Organization and properties of DHHC palmitoyltransferases (PATs)
Click image to view.
a. The organization of three different DHHC-PATs are shown schematically. The spheres indicate protein-protein interaction domains. Erf2 associates with a cytoplasmic subunit, Erf4, to form the active enzyme.
b. The DHHC-CRD region of a few representative DHHC-PATs are aligned. The conserved amino acids are shown in red.
c. Reaction catalyzed by DHHC-PATs; the reverse reaction is catalyzed by acylprotein thioesterases (APT).
Besides its broad importance in cell biology, palmitoylation has been linked to several diseases, most notably neuropsychiatric disorders such as Huntington’s disease and various forms of cancer. Recently, it was shown that zDHHC20 palmitoylates epidermal growth factor (EGFR) and is thus a potential therapeutic target for a wide range of cancers. More recently, zDHHC3 has been proposed as a target for cancer treatment owing to its activity as the palmitoyltransferase for the programmed cell-death ligand 1 (PD-L1). However, when we started working on this family, very little was known about the molecular mechanism of zDHHC palmitoyltransferases, despite their importance across a broad spectrum of biological pathways and their biomedical importance. Nothing was known about their structural organization or how they interact with substrates and the fatty acyl coenzyme A (CoA), which serves as the acyl donor.
In a major breakthrough in this field, we had earlier solved the high-resolution crystal structures of two members of the zDHHC family, human zDHHC20 (hDHHC20) and zebrafish zDHHC15 (Figure 2a), the first structures of any member of this family to be characterized. They reveal a tepee-like transmembrane domain organization, which splays apart towards the cytoplasmic side and harbors the active site at the membrane-aqueous interfacial region (Figure 2b), thus readily explaining why membrane-proximal cysteines are palmitoylated. We also solved the structure of hDHHC20 irreversibly modified by a covalent inhibitor, 2-bromopalmitate. The structure mimics the auto-acylated intermediate state in the enzymatic pathway and thus reveals how the acyl group of fatty acyl–CoA binds in a cavity formed in the bilayer by the transmembrane domain (Figure 2c). Residues lining the cavity contact the acyl chain, and their mutation affects enzymatic activity. By mutating two residues at the tapering end of the cavity, we also showed that we can change the acyl chain–length selectivity of the mutant enzymes (Figure 2d). Thus, the cavity functions as a molecular ruler in determining the acyl chain–length selectivity of hDHHC20, important because, although palmitate is the most prevalent fatty acid used by DHHC palmitoyltransferases, they can use fatty acyl–CoAs of other chain lengths, a property that varies between different members of the DHHC family.
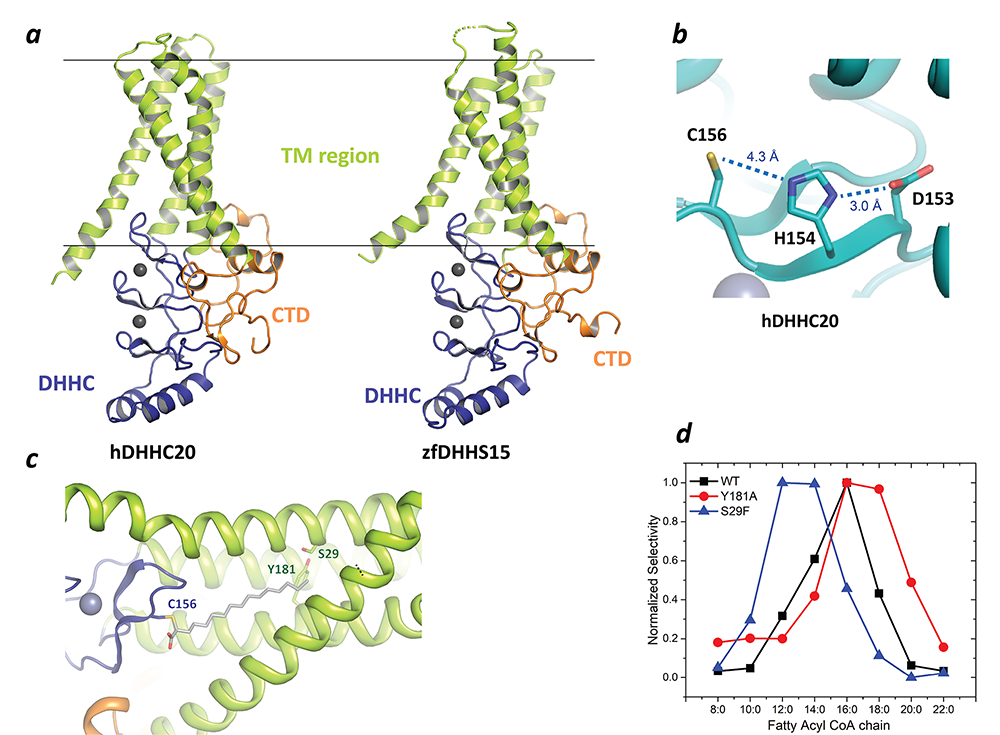
Figure 2. Structure, function, and membrane deformation of DHHC palmitoyltransferases
Click image to view.
a. The structure of human DHHC20 and a catalytically inactive mutant of zebrafish DHHC15, shown in ribbon trace. The transmembrane domain (TM) is shown in green, the DHHC–containing cysteine-rich domain in blue and the C-terminal domain in orange. The grey spheres indicate Zn2+ ions. These are both Golgi-resident enzymes, and thus the top side faces the Golgi lumen and the active site the cytoplasm.
b. Active site of human DHHC20 showing the catalytic triad containing the active-site cysteine.
c. Structure of human DHHC20 irreversibly modified with 2-bromopalmitate, which results in the active-site cysteine linking to the alpha-carbon of palmitic acid. The acyl group of palmitic acid is shown in stick rendition. Also shown are two residues towards the top of the tapering cavity to where the palmitate binds.
d. The acyl chain–length selectivity of wild-type (WT) human DHHC20. Mutation of tyrosine181 to alanine (Y181A) expands the cavity and shifts the acyl selectivity to the longer side. On the other hand, mutation of serine29 to phenylalanine (S29F) contracts the cavity and thus shifts the acyl selectivity to the shorter side.
S-acylation was first discovered in viral proteins and in the wake of the COVID-19 pandemic, so we turned our attention to this. It had been shown that the Spike protein of other coronaviruses or its equivalent in other pathogenic viruses are S-acylated and, in a subset of these, S-acylation is crucial to the viral life cycle. However, it was not known whether the Spike protein of SARS-CoV-2, the causative agent of COVID-19, is S-acylated and, if so, how that affects the viral life cycle. Remarkably, even 40 years after the discovery of S-acylation, there were still no in vitro reconstitution of S-acylation of any viral protein with purified components. We started a project approved by the NIH COVID-19 committee to answer these questions. Our experiments demonstrated, using a click chemistry–based in cellulo assay, that the Spike protein of SARS-CoV-2 is S-acylated (Figure 3). We went on to identify specific cysteine clusters that are targets of S-acylation. Interestingly, when we investigated the effect of the cysteine clusters using pseudotyped virus, in collaboration with Eric Freed's lab, mutation of the same three clusters of cysteines (that are S-acylated) severely compromised viral infectivity. We developed a library of expression constructs of human zDHHC enzymes and used them to identify zDHHC enzymes that can S-acylate the SARS-CoV-2 Spike protein. We reconstituted S-acylation of the SARS-CoV-2 Spike protein in vitro using purified zDHHC enzymes and observed a striking heterogeneity in the S-acylation status of the different cysteines in our in cellulo experiments, which, remarkably, was recapitulated by the in vitro assay. Taken together, these results bolster our understanding of a poorly understood post-translational modification integral to the SARS-CoV-2 Spike protein. The study also opens up avenues for further mechanistic dissection and lays the groundwork for developing future strategies that could aid in the identification of targeted small-molecule modulators.
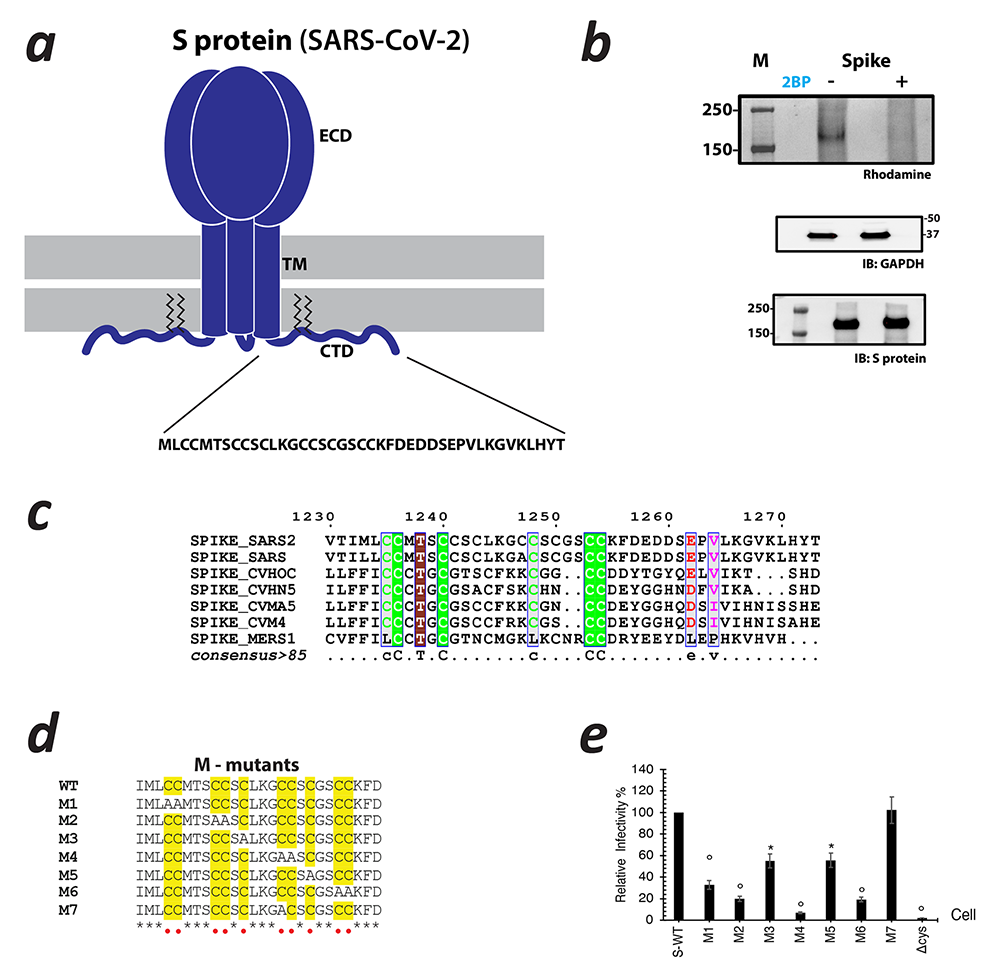
Figure 3. S-acylation of the SARS-CoV-2 Spike protein
Click image to view.
a. Schematic showing the trimeric form of SARS-CoV-2 Spike protein with the ectodomain (ECD), Transmembrane (TM) region, and C-terminal Domain (CTD). The CTD is further enhanced to show the cystine clusters, which are potential sites of S-acylation.
b. S-acylation of the Spike protein determined by click chemistry. Gel images detecting rhodamine signal at the position of Spike protein in the presence and absence of 2-bromopalmitate (2BP), a global inhibitor of S-acylation. Western blots probed with antibodies against GAPDH and the Spike protein respectively serve as controls for loading and total Spike protein expression.
c. Sequence alignment of the CTDs of Spike protein from various coronaviruses.
d. Sequence alignment showing Spike protein mutants. Cysteine residues are highlighted in yellow. They are built on the backbone of wild-type (WT) Spike protein, M1 through M6 indicating either an individual cysteine or di-cysteine motif that is mutated to alanine. The M7 mutant represents a construct in which the additional cysteine in SARS-CoV-2 with respect to SARS-Co-V has been mutated out.
e. Infectivity of HIV-1 particles pseudotyped with WT Spike protein and the cysteine mutants.
Molecular mechanism of iron and polyamine transport across cellular membranes
The importance of iron in biology cannot be overstated. In higher organisms, mitochondria are the ‘hotspot’ for the cell biology of iron, because Fe-S clusters are biosynthesized and iron is inserted into heme there. Mitochondrial iron homeostasis plays a critical role in cellular iron homeostasis and in the overall physiology of the cell. In vertebrates, the only known major transporters of iron into mitochondria are mitoferrin-1 and mitoferrin-2, two homologous members of a large group of mitochondrial transporters known as the Mitochondrial Carrier family (Figure 4). Mitoferrin-1 (Mfrn1) is expressed mainly in erythroid cells, while mitoferrin-2 is expressed ubiquitously. Knockout of Mfrn1 is embryonically lethal, reflecting the importance of mitoferrins in vertebrate physiology.
Mfrn1 and Mfrn2 were discovered more than 15 years ago. However, the proposed iron-transport activity had not been demonstrated using an in vitro functional reconstitution assay, and nothing was known about their interaction with iron or other related metal ions, most likely because heterologous overexpression and purification of mitoferrins were not reported in the literature. There were also no reports of a reconstituted iron transport assay starting from purified protein. In earlier work, we carried out heterologous purification, in vitro functional reconstitution, and mutational dissection of a vertebrate Mfrn1 (Figure 4), the first demonstration that Mfrn1 can indeed transport iron. Our studies provided the first biochemical insights into Mfrn function. In earlier work, we also used our in vitro proteoliposome-reconstituted iron-transport assay, the first such assay to be reported in the literature, to dissect the iron-transport activity of MavN, another highly conserved iron transporter, in the bacterial pathogen Legionella pneumophila (Figure 4b).
In the past year we shifted our interest to transporters that move polyamines across the membrane. Polyamines, small organic polycations, are essential for cell viability, and their physiological levels are homeostatically maintained by post-transcriptional regulation of key biosynthetic enzymes. In addition to de novo synthesis, cells can also take up polyamines; however, until recently, dedicated polyamine transporters were not known. Tom Dever's lab discovered that the S. cerevisiae HOL1 mRNA is under translational control by polyamines. They also showed that Hol1 is required for yeast growth under limiting polyamine conditions. These experiments suggested that Hol1 is a high-affinity polyamine transporter (Figure 4c). We purified recombinant Hol1 and reconstituted it in protealiposomes to demonstrate that it indeed shows robust polyamine transport activity. An extensive set of experiments from the Dever lab bolstered the conclusions that Hol1 is a high-affinity polyamine transporter and under translational autoregulation by polyamines in yeast, highlighting the extensive control cells impose on polyamine levels. We are now investigating the underlying molecular mechanisms behind Hol1 function.
Click image to view.
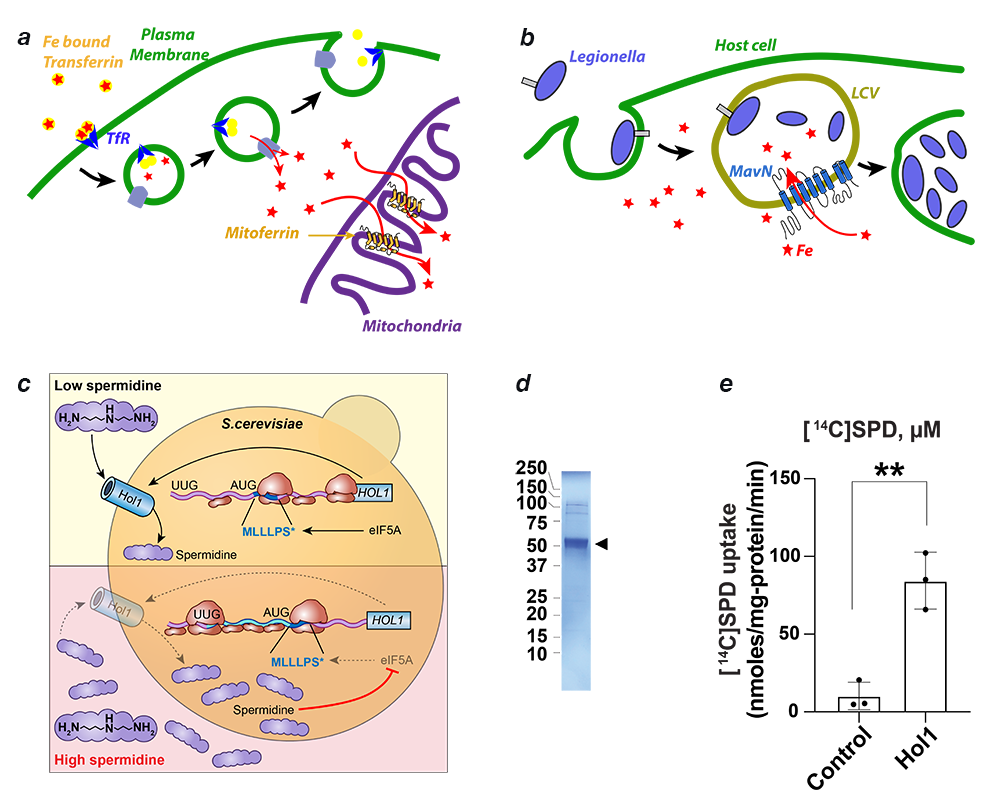
Figure 4. Transporters of iron and polyamine
a. Iron is imported through the plasma membrane by the transferrin/transferrin receptor (blue) cycle and is transported out of endosomes by the divalent metal ion transporter (DMT) (grey). Iron is delivered to mitoferrin (yellow cylinders) by unknown means. Mitoferrin delivers iron to unknown partners in mitochondria, which become available for heme and Fe-S cluster biosynthesis.
b. Schematic showing Legionella entering a host cell and sequestering itself in a Legionella-containing vacuole (LCV). MavN is inserted into the membrane of the LCV and hijacks iron from the host cell.
c. Assay showing iron transport by proteoliposome-reconstituted MavN. Hol1 as the major high-affinity polyamine transporter in S. cerevisiae. Polyamines autoregulate HOL1 expression through feedback inhibition of HOL1 mRNA translation mediated by polyamine inhibition of eIF5A–dependent translation termination on a conserved upstream open reading frame in the HOL1 mRNA leader.
d. SDS-PAGE gel of detergent-purified K. lactis Hol1 (arrowhead).
e. [14C]Spermidine uptake by control or K. lactis Hol1 proteoliposomes. Error bars denote SD, **p < 0.01 (Student’s two-tailed t test, n = 3).
Structure and molecular mechanism of ATG9, the only transmembrane component of the core autophagy machinery
Autophagy is a critical process in both health and disease. Serving mainly to adapt organisms to a diverse range of conditions such as metabolic stress, pathogenic infection, and ageing, autophagy is conserved from yeast to humans. In addition, autophagy is involved in a large number of human diseases, including neuro-degenerative disorders, various forms of cancer, and inflammatory diseases. Although autophagy was discovered more than 50 years ago, some its fundamental aspects remain rather poorly understood and are only now coming to light. An outstanding example of this lack of knowledge is the structure and function of ATG9, the only essential transmembrane protein in the pathway. ATG9 localizes to small, 30–60 nm vesicles known as 'ATG9 vesicles,' which are critically important for the expansion of the autophagosome membrane. ATG9 vesicles traffic between the trans-Golgi network (TGN) and pre-autophagosomal structures in response to stimuli that initiate autophagy. Despite the clear and obvious importance of ATG9 in autophagy, its atomic structure and molecular function remain unknown. It has been speculated that ATG9 transports lipids that contribute to the growth of the double-membrane structure of the autophagosome. However, progress in this regard has been completely thwarted by the lack of an atomic structure that could serve as a starting point for a range of experiments, from cell-biological to biochemical, to elucidate its function.
In order to address this knowledge gap, we initiated a collaboration with the labs of Juan Bonifacino and Jiansen Jiang. This led to a 2.9 Å–resolution structure of the ubiquitous human ATG9A isoform by cryo-EM (Figure 5a). The structure revealed that ATG9A adopts a unique fold (Figure 5b, 5c), assembling into a domain-swapped homotrimer, with each wedge-shaped protomer comprising four transmembrane α-helices and two α-helices that are only partially embedded in the membrane. The transmembrane domain is capped by a cytosolic domain, which is mostly α-helical. Classification of the cryo-EM imaging revealed two predominant ATG9A conformers (states A and B). An important feature of the ATG9A structure is the presence of a branched network of pores through the protein, with outlets facing the cytosol, the outer leaflet lipid head groups, and the lumen (Figure 5d). Cell-based functional assays demonstrated the importance of pore-lining residues, suggesting an essential function of the pore. The dimensions of the pore are sufficient to accommodate phospholipids and likely permit lipid transport from or to a lipid chaperone such as ATG2. Alternatively ATG9 may act as a lipid scramblase, distributing the lipids that arrive through ATG2 among two leaflets of the bilayer. The branched pores may also function as solvent conduits to relieve osmotic pressures in growing and shrinking ATG9 vesicles. ATG9A directly interacts with ATG2A, interactions that are mediated by the ATG9A cytosolic domain, deletion of which abrogates both ATG2A co-immunoprecipitation and autophagosome maturation. Because ATG2A has demonstrated lipid-transport capability in vitro, the plausible functions of ATG9A are to: (1) facilitate movement of lipids across the bilayer of the growing phagophore, which could be achieved by using the branched pores we see in our structure for moving polar lipid headgroups or by distorting the bilayer, lowering the energetic barrier for trans-bilayer lipid movement; (2) act as a membrane-anchored 'lasso' to capture and accurately target ATG2A or other lipid chaperones. It is interesting to note that two helices, which plug the central pore of the trimer, are discernible only in state A, indicating that flexibility of the putative 'lasso' could be partly dependent on the conformation of the transmembrane core.
The early stages of autophagy involve a reorganization of intracellular membranes to form the nascent autophagosome. ATG9 is essential for this process, and our structure provides a framework for interrogating its function in bilayer remodeling. Using molecular dynamics simulations in collaboration with the lab of José Faraldo-Gómez, and based on the cryo-EM data, we found that the architecture of the ATG9A trimer elicits a long-range positive membrane curvature, owing to the geometric features and amino-acid make-up of the protein surface exposed to the lipid. The simulations also show that co-localization of multiple ATG9A trimers greatly amplifies this effect and induces macroscopic changes in membrane morphology (Figure 5e, 5f). The finding corresponds with observations that ATG9 is present in small (30–60 nm) vesicles and thin tubules, and, at least in yeast, localizes to the edges of the growing phagophore. Interestingly, states A and B differ in the tilt-angle of the protomers relative to the membrane normal, implying a tolerance for a range of bilayer curvatures. This leads to the intriguing possibility that ATG9A–induced curvature may modulate the energetic scale and effect lipid diffusion out of the vesicles into another membrane.
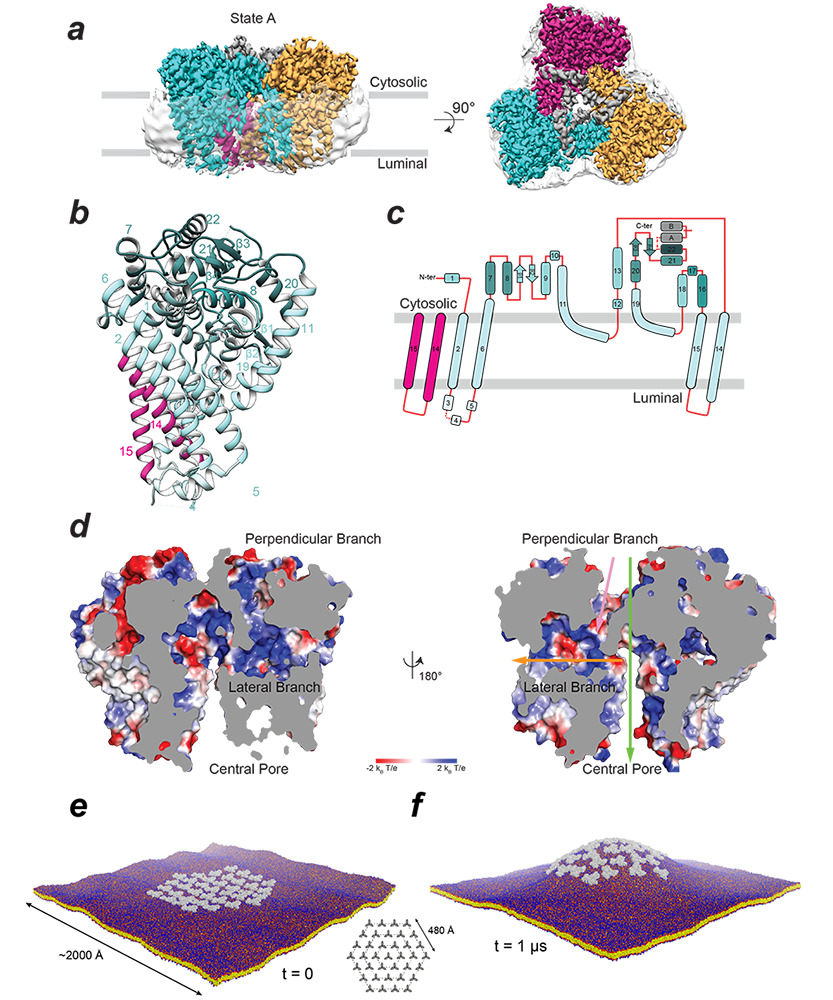
Click image to view.
Figure 5. Structure and membrane interactions of human ATG9A
a. Cryo-EM density map of ATG9A in state A is shown in side and top views and colored according to the protomer (cyan, magenta, and orange). Translucent surface shows disordered detergent molecules surrounding the transmembrane surface of the trimer.
b. Structure of an isolated protomer (cyan) and two alpha helices from another protomer (magenta). Numbers correspond to alpha helices.
c. Topology of the ATG9A protomer showing numbered alpha helices as rounded rectangles and beta strands as filled arrows.
d. Branched network of internal pores in the ATG9A trimer shown by electrostatic potential surface of the central pore, lateral branch, and perpendicular branch (with 180° rotation). Green, yellow, and pink arrows are used to show the positions of the central pore, lateral branch, and perpendicular branch, respectively.
e. and f. Molecular dynamics simulation of a hypothetical ATG9A cluster in a POPC (2-oleoyl-1-pamlitoyl-sn-glyecro-3-phosphocholine) bilayer.
e. Configuration of the system at the beginning of the simulation; 36 ATG9A proteins (gray) are arranged around a central trimer in three concentric hexagons (inset). Note the proteins are not in contact. The system amounts to about 11,220,000 particles.
f. At the end of the calculated trajectory of 1 ms.
Publications
- Puthenveetil R, Lun CM, Murphy RE, Healy LB, Vilmen G, Christenson ET, Freed EO, Banerjee A. S-acylation of SARS-CoV-2 spike protein: mechanistic dissection, in vitro reconstitution and role in viral infectivity. J Biol Chem 2021;297(4):101112–101124.
- Vindu A, Shin BS, Choi K, Christenson ET, Ivanov IP, Cao C, Banerjee A, Dever TE. Translational autoregulation of the S. cerevisiae high-affinity polyamine transporter Hol1. Mol Cell 2021;81(19):3904–3918.
- Vilmen G, Banerjee A, Freed EO. Rafting through the palms: S-acylation of SARS-CoV-2 spike protein induces lipid reorganization. Dev Cell 2021;56(20):2787–2789.
- Guardia CM, Christenson ET, Zhou W, Tan XF, Lian T, Faraldo-Gómez JD, Bonifacino JS, Jiang J, Banerjee A. The structure of human ATG9A and its interplay with the lipid bilayer. Autophagy 2020;16(12):2292–2293.
Collaborators
- Juan Bonifacino, PhD, Section on Intracellular Protein Trafficking, NICHD, Bethesda, MD
- David Cafiso, PhD, Department of Chemistry, University of Virginia, Charlottesville, VA
- Thomas Dever, PhD, Section on Mechanism and Regulation of Eukaryotic Protein Synthesis, NICHD, Bethesda, MD
- José Faraldo-Gómez, PhD, Theoretical Molecular Biophysics Section, NHLBI, Bethesda, MD
- Eric Freed, PhD, Retroviral Replication Laboratory, NCI, Frederick, MD
- Rodolfo Ghirlando, PhD, Laboratory of Molecular Biology, NIDDK, Bethesda, MD
- James Inglese, PhD, Assay Development & Screening Technology Laboratory, NCATS, Bethesda, MD
- Ralph Isberg, PhD, Tufts University School of Medicine, Boston, MA
- Jiansen Jiang, PhD, Laboratory of Membrane Proteins and Structural Biology, NHLBI, Bethesda, MD
Contact
For more information, email anirban.banerjee@nih.gov or visit https://banerjee.nichd.nih.gov.