Childhood Neurodegenerative Lysosomal Storage Disorders

- Anil B. Mukherjee, MD, PhD, Head, Section on Developmental Genetics
- Maria B. Bagh, PhD, Research Fellow
- Abhilash Appu, PhD, Visiting Fellow
- Avisek Mondal, PhD, Visiting Fellow
- Nisha Plavelil, PhD, Visiting Fellow
- Tamal Sadhukhan, PhD, Visiting Fellow
- Koyel Roy, MSc, Contract Biologist
- Sriparna B. Sadhukhan, MSc, Contract Biologist
The Section on Developmental Genetics conducts both basic and translational research into a group of the most common childhood neurodegenerative lysosomal storage disorders (LSDs), called neuronal ceroid lipofuscinoses (NCLs), commonly known as Batten disease. These diseases mostly affect children, and there is no curative treatment for any of the NCLs. Mutations in at least 14 different genes (called CLNs) underlie various forms of NCL. The CLN1, CLN2, CLN5, CLN10, and CLN13 genes encode soluble lysosomal enzymes; the CLN4 and CLN14 encode peripherally associated cytoplasmic proteins; the CLN11 encodes progranulin, a protein in the secretory pathway; and several transmembrane proteins with various subcellular localizations are encoded by CLN3, CLN6, CLN7, CLN8, and CLN12. The infantile NCL (INCL), a fatal neurodegenerative LSD, is caused by inactivating mutations in the CLN1 gene. CLN1 encodes a lysosomal depalmitoylating enzyme called palmitoyl-protein-thioesterase-1 (PPT1). Our investigations focus on understanding the molecular mechanisms of pathogenesis underlying INCL (CLN1 disease), juvenile NCL (JNCL: CLN3 disease), and congenital NCL (CNCL: CLN10 disease). Interestingly, all NCL types share some common clinical features such as epileptic seizures, progressive psychomotor decline, and visual impairment resulting from retinal degeneration. The pathologic features include intracellular accumulation of auto-fluorescent material, neuro-inflammation, and cortical atrophy. Such patients also have a shortened lifespan.
Several years ago, we initiated investigations on INCL. Numerous proteins, especially in the brain, require S-palmitoylation (also called S-acylation), a reversible post-translational modification in which a 16-carbon, saturated fatty acid (generally palmitic acid) is attached to specific cysteine residues in polypeptides via thioester linkage. While S-palmitoylation plays important roles in membrane anchorage of soluble proteins, protein-protein interactions, protein trafficking, and protein stability, such lipid-modified proteins must also be depalmitoylated for recycling or degradation and clearance by lysosomal hydrolases. Thus, dynamic S-palmitoylation (palmitoylation-depalmitoylation), just as phosphorylation-dephosphorylation, regulates the function of many proteins, especially in the brain. Dynamic S-palmitoylation requires coordinated actions of two types of enzyme with opposing functions. The enzymes that catalyze S-palmitoylation are palmitoyl acyltransferases (PATs), which are zinc-finger proteins with a common DHHC (Asp-His-His-Cys) motif, and they are called ZDHHC PATs or simply ZDHHCs. The mammalian genome encodes a family of 23 ZDHHC PATS. Similarly, the palmitoyl thioesterases, which depalmitoylate S-acylated proteins, are localized either in the lysosomes like PPT1 or in the cytoplasm like acyl-protein thioesterase-1 (APT1). Recently, several protein depalmitoylases called ABHD17 were identified, which catalyze the turnover of N-Ras (a GTP-ase signal-transduction protein).
PPT1 catalyzes the cleavage of the thioester linkage of S-palmitoylated proteins, which is vitally important because such lipid-modified proteins are refractory to degradation by lysosomal hydrolases. Thus, PPT1 deficiency leads to lysosomal accumulation of the S-palmitoylated proteins (constituents of ceroid), which has been proposed as the mechanism of INCL pathogenesis. However, the precise molecular mechanism underlying INCL pathogenesis has remained elusive for more than two decades. Children afflicted with INCL are normal at birth but, by 11 to 18 months of age, exhibit signs of psychomotor retardation. By two years of age, they are completely blind owing to retinal degeneration and, by age four, they manifest no brain activity and remain in a vegetative state for several more years before eventual death. Such grim outcomes underscore the urgent need for the development of rational and effective therapeutic strategies, not only for INCL but also for all NCLs.
The aim of our translational research is to apply the knowledge gained from our basic laboratory investigations to develop novel therapeutic strategies for Batten disease. The results of our earlier investigations on INCL led to a bench-to-bedside clinical trial. Using Cln1–knockout (Cln1–/–) mice, which recapitulate virtually all clinical and pathological features of INCL, we discovered that PPT1 deficiency causes endoplasmic-reticulum (ER) and oxidative stress, which at least in part causes neuronal death by apoptosis. During the past several years, we also delineated a mechanism by which PPT1 deficiency disrupts the recycling of synaptic-vesicle (SV) proteins, which are essential for generating fresh SVs to replenish the SV pool size at the nerve terminals so as to maintain uninterrupted neurotransmission. We also discovered that ER and oxidative stress contribute to neuronal apoptosis and neuro-inflammation in INCL. Further, we found that PPT1 deficiency causes mis-routing of the V0a1 subunit of v-ATPase (the proton pump on lysosomal membrane), which dysregulates lysosomal acidification, causing elevated pH and thus adversely affecting lysosomal degradative function.
We also developed a non-invasive method, using MRI and MRS (magnetic resonance spectroscopy) to evaluate the progression of neurodegeneration in Cln1–/– mice. The methods permit repeated evaluation of potential therapeutic agents in treated animals. Application of such methods in our clinical trial with INCL also allowed us to evaluate the progressive decline in brain volume and neurodegeneration. In collaboration with Wadih Zein, we are also conducting studies to determine whether electro-retinography can be used to assess the progressive retinal deterioration in Cln1–/– as well as in Cln1–knockin (KI) mice, which carry the nonsense mutation in the CLN1 gene commonly found in the INCL patient population in the US. Moreover, we discovered that the blood-brain barrier is disrupted in Cln1–/– mice and that this pathology is ameliorated by treatment with resveratrol, which has antioxidant properties. More recently, we discovered that a nucleophilic small molecule with antioxidant properties, N-(tert-butyl) hydroxylamine (NtBuHA), ameliorates the neurological abnormalities in Cln1–/– mice and extends their lifespan. The compound is currently undergoing preclinical evaluation for the approval of an IND by the FDA. Intriguingly, we discovered that in Cln1–/– mice the lysosomes contain insufficient amounts of PPT1 protein and PPT1–enzymatic activity, contributing to neuro-pathology in this disease. These and related studies provide insight into the complex mechanisms of heritable disorders of neurodegeneration such as CLN1 disease (INCL) as well as CLN3 disease (JNCL) and identify several potential therapeutic targets. Our results suggest that thioesterase-mimetic small molecules such as NtBuHA are potential therapeutics for INCL and may even be for JNCL. More recently, we discovered that cathepsin D (CD) deficiency in lysosomes is a common pathogenic link between CLN1 disease and CLN10 disease (CNCL). Our ongoing laboratory and translational investigations are attempting to advance our knowledge of INCL, JNCL, and congenital NCL (CNCL) diseases.
Figure 1. Dysregulation of lysosomal calcium homeostasis in a mouse model of INCL
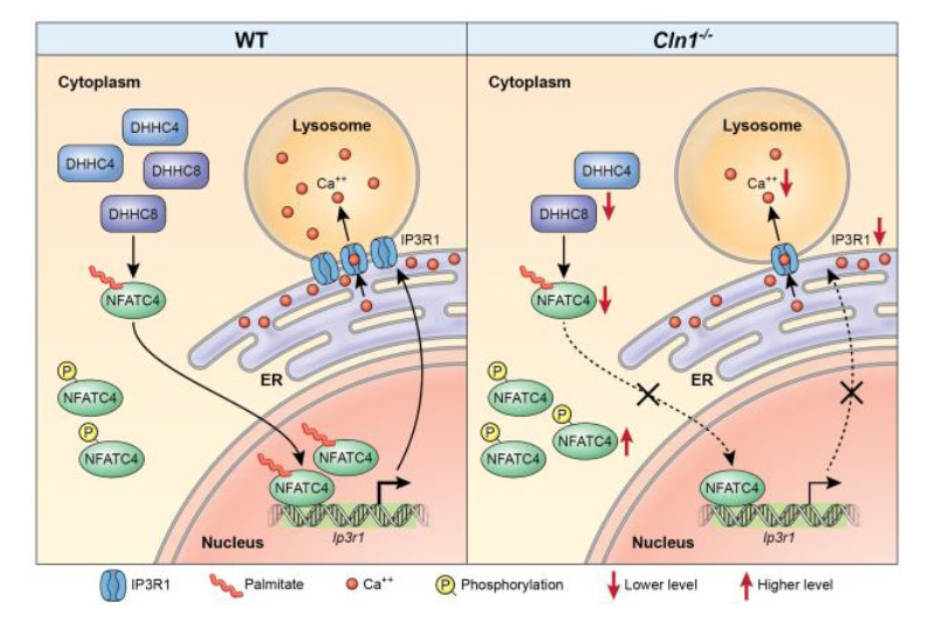
Click image to view.
Schematic representation showing how S-palmitoylation of NFATC4 may regulate IP3R1 expression thereby controlling lysosomal Ca2+-homeostasis, which is dysregulated in INCL mice. In the Cln1–/– mouse brain, the levels of two palmitoyl acyl transferase enzymes (i.e., ZDHHC4 and ZDHHC8), enzymes that catalyze S-palmitoylation of NFATC4, are reduced. As a result of their reduced levels in Cln1–/– mouse brain, S-palmitoylation of NFATC4 is suppressed, preventing its trafficking to the nucleus, which dysregulates Ip3r1 expression, thereby, suppressing the transport of Ca2+ from the ER to the lysosome. Low lysosomal Ca2+ suppresses the catalytic activities of Ca2+–dependent lysosomal acid hydrolases, causing storage of S-acylated proteins in lysosomes, which contributes to INCL pathogenesis.
Cln3 mutations significantly reduce lysosomal Ppt1–protein and Ppt1–enzyme activity.
Because PPT1 deficiency causes lysosomal accumulation of autofluorescent ceroid, leading to INCL, and intracellular accumulation of ceroid is a characteristic of all NCLs, a common pathogenic link for these diseases has been suggested. It has been reported that CLN3 mutations suppress the exit of the cation-independent mannose 6-phosphate receptor (CI-M6PR) from the trans-Golgi network (TGN). Because CI-M6PR transports soluble proteins such as PPT1 from the TGN to the lysosome, we hypothesized that CLN3 mutations may cause lysosomal PPT1 insufficiency, thus contributing to JNCL pathogenesis. We found that the lysosomes in Cln3–mutant mice, which mimic JNCL, and those in cultured cells from JNCL patients, contain significantly reduced levels of the Ppt1 protein and Ppt1–enzyme activity and progressively accumulate autofluorescent ceroid. Furthermore, in JNCL fibroblasts the V0a1 subunit of v-ATPase, which regulates lysosomal acidification, is mis-localized to the plasma membrane instead of to its normal location on lysosomal membrane. The defect dysregulates lysosomal acidification, as we previously reported in Cln1–/– mice. Our findings uncover a previously unrecognized role of CLN3 in lysosomal homeostasis and suggest that CLN3 mutations causing lysosomal Ppt1 insufficiency may at least in part contribute to JNCL pathogenesis.
Impaired lysosomal Ca2+ homeostasis contributes to pathogenesis in INCL mice.
The lysosome is an organelle long known for mediating degradation and clearance of cellular waste. In recent years, it has become evident that it is a highly dynamic structure that also plays important roles in cell metabolism in response to environmental cues. Impaired lysosomal degradative function leads to a family of about 60 inherited LSDs. Dysregulation of cellular Ca2+ homeostasis is reported to play important roles in the pathogenesis of several human diseases, including the LSDs. Defective lysosomal Ca2+ homeostasis has also been reported to impair autophagy. In most of the LSDs, defective autophagy leads to neurodegeneration.
The ER is the major Ca2+repository in the cell, and Ca2+ plays a key regulatory role in autophagy. It is an intracellular degradative process that requires Ca2+–dependent lysosomal hydrolases for the degradation and clearance of the cargo contained in the autophagosomes. Lysosomal Ca2+homeostasis is mediated by inositol 3-phosphate receptor 1(IP3R1)–mediated transport of Ca2+ from the ER to the lysosome. It has also been reported that selective interaction of IP3Rs with the ER–lysosome contact sites is required for the delivery of Ca2+ to the lysosome. Moreover, antagonists of IP3Rs rapidly and completely block lysosomal Ca2+ refilling. Interestingly, IP3R1 has been reported to undergo S-palmitoylation for regulating Ca2+ flux in immune cells. Furthermore, disruption of Ca2+ homeostasis may dysregulate neurotransmitter release, contributing to neurodegeneration. Autophagy is impaired by dysregulation of Ca2+homeostasis in many LSDs including in Cln1–/– mice. We sought to test the hypothesis that CLN1 mutations dysregulate lysosomal Ca2+ homeostasis and suppress the catalytic activities of Ca2+–dependent lysosomal hydrolases, which impair the degradation of undigested cargo in autophagosomes, causing neuro-pathology in INCL.
We sought to determine the mechanism by which PPT1 deficiency impairs lysosomal degradative function and contributes to INCL pathogenesis. We found that in Cln1–/– mice low levels of IP3R1 dysregulate lysosomal Ca2+ homeostasis. Intriguingly, the transcription factor NFATC4, which regulates IP3R1 expression, required S-palmitoylation for trafficking from the cytoplasm to the nucleus. We identified two palmitoyl acyltransferases, ZDHHC4 and ZDHHC8, which catalyzed S-palmitoylation of NFATC4. Notably, in Cln1–/–mice, reduced ZDHHC4 and ZDHHC8 levels markedly lowered S-palmitoylated NFATC4 (active) in the nucleus, which inhibited IP3R1 expression, thereby, dysregulating lysosomal Ca2+ homeostasis. Consequently, Ca2+–dependent lysosomal enzyme activities were markedly suppressed. Impaired lysosomal degradative function impaired autophagy, which caused lysosomal storage of undigested cargo. Importantly, IP3R1 overexpression in Cln1–/– mouse fibroblasts ameliorated this defect. Our results reveal a previously unrecognized role of Cln1/Ppt1 in regulating lysosomal Ca2+ homeostasis and suggest that the defect contributes to INCL pathogenesis.
Dysregulation of lysosomal acidification contributes to neurodegeneration in INCL mice.
In eukaryotic organisms, the lysosome is the primary organelle for intracellular digestion. It contains enzymes that require an acidic pH for optimal degradative function. Thus, lysosomal acidification is of fundamental importance for the degradation of macromolecules of intra- and extracellular origin that are delivered to the lysosome, and its dysregulation contributes to pathogenesis in virtually all LSDs, including NCLs. Furthermore, defective regulation of lysosomal pH has also been reported in common neurodegenerative diseases such as Alzheimer’s and Parkinson’s. However, despite intense studies, the precise mechanism(s) underlying defective lysosomal acidification in these diseases has remained elusive. Lysosomal acidification is regulated by vacuolar ATPase (v-ATPase), a multi-subunit protein complex consisting of the cytosolic V1 sector and the lysosomal membrane–anchored V0-sector. Reversible assembly of V1/V0 sectors on the lysosomal membrane maintains functionally active v-ATPase, the proton pump of the cell, which regulates lysosomal acidification.
Two thioesterases are cytosolic (APT1 and APT2) and two (PPT1 and PPT2) are localized to the lysosome. Dynamic S-palmitoylation (palmitoylation–depalmitoylation) requires the coordinated action of these two groups of enzymes with opposing functions (i.e., ZDHHCs and PPTs), which maintains the steady-state membrane localization and function of numerous important proteins, especially in the brain. By catalyzing depalmitoylation, thioesterases also facilitate recycling or degradation of proteins that undergo S-palmitoylation.
We tested a hypothesis that one or more subunits of v-ATPase require S-palmitoylation for endosomal sorting, trafficking, and reversible assembly of V1/V0 on the lysosomal membrane, which is essential for regulating lysosomal pH, and that Ppt1 deficiency disrupts v-ATPase activity, impairing its proton-transport function, thereby dysregulating acidification of lysosomal lumen. Our results show that the lysosomal membrane–anchored V0 sector isoform a1 (V0a1) subunit of v-ATPase indeed undergoes S-palmitoylation, which is required for its sorting and trafficking to the lysosomal membrane. The process appears to be defective in Ppt1–deficient Cln1–/– mice. Notably, we demonstrated that treatment of these mice with the thioesterase (Ppt1)–mimetic small molecule NtBuHA restores near normal v-ATPase activity and rescues the defective lysosomal acidification phenotype. The results demonstrate the potential of NtBuHA as a drug target for INCL.
Cholesterol- and IGF1–mediated mTORC1 signaling contribute to INCL neuro-pathology.
The lysosome has long been considered the terminal organelle for degradation. However, emerging evidence has, remarkably, changed our understanding of the lysosome as a terminal degradative organelle to a critical mediator of fundamental metabolic processes. Signals from nutrients such as glucose, amino acids, fatty acids, and cholesterol are integrated by the lysosome, turning the cellular events from anabolic to catabolic processes, including autophagy. Whereas materials from extracellular sources are transported to the lysosome via endocytosis, materials originating from intracellular sources are delivered by autophagy. Autophagy is a critical process, dysregulation of which is implicated in many LSDs. It has been suggested that the autophagy-lysosomal pathway plays critical roles in many cellular functions, including signaling in response to environmental cues. Dysregulation of autophagy also underlies pathogenesis of common neurodegenerative diseases such Alzheimer’s and Parkinson’s. There are three types of autophagy: macroautophagy, microautophagy, and chaperone-mediated autophagy. In all three types the lysosome plays a pivotal role in the degradation of cargo contained in autophagosomes.
The mechanism(s) underlying neurodegeneration in various LSDs has not been clearly defined. The mechanistic target of rapamycin (mTOR), a serine/threonine kinase, is a master regulator of cellular growth and metabolism. It is activated by cues from nutrients and growth factors as well as by cholesterol. Activation of mTOR signaling, which impairs autophagy, underlies both neuro-psychiatric and neurodegenerative disorders. We sought to determine whether Ppt1 deficiency in Cln1–/– mice mediates aberrant activation of mTORC1 signaling, suppressing autophagy and contributing to neuro-pathology in this mouse model of INCL. We found that several constituent proteins (e.g., vATPase and Lamtor1) of the lysosomal nutrient-sensing scaffold (LNSS) and the lysosomal cholesterol exporter, Niemann-Pick Complex 1 (NPC1) protein, require S-palmitoylation for lysosomal targeting. However, in Cln1–/– mice, lack of Ppt1 caused mis-targeting of these proteins to the plasma membrane instead of their normal location on lysosomal membrane. We found that, despite the disruption of the LNSS, aberrant activation of mTORC1 signaling occurred via insulin-like growth factor 1 (IGF1)– and cholesterol-mediated pathways. Importantly, pharmacological inhibition of these pathways significantly suppressed mTORC1 signaling and ameliorated the dysregulation of autophagy, implicating these agents as potential drug targets for INCL.
Ablation of microRNA-155 does not ameliorate neuro-inflammation in INCL mice.
The INCL–mimicking Cln1–/– mice manifest progressive neuro-inflammation, contributing to neurodegeneration. However, the underlying mechanism of neuro-inflammation in INCL and in Cln1–/– mice has remained elusive. Previously, it had been reported that microRNA-155 (miR-155) regulates inflammation, and miR profiling in the Cln1–/– mouse brain showed that the level of miR-155 was upregulated. Thus, we sought to determine whether ablation of miR-155 in Cln1–/– mice suppresses neuro-inflammation in such mice. Towards this goal, we generated Cln1–/–/miR-155–/– double knockout (KO) mice and evaluated the inflammatory signatures in the brain. We found that the brains of double KO mice manifest progressive neuro-inflammatory changes virtually identical to those found in Cln1–/– mice. We conclude that ablation of miR-155 in Cln1–/– mice does not alter the neuro-inflammatory trajectory in INCL mouse model.
Publications
- Appu AP, Bagh MB, Sadhukhan T, Mondal A, Casey S, Mukherjee AB. Cln3-mutations underlying juvenile neuronal ceroid lipofuscinosis cause significantly reduced levels of palmitoyl-protein thioesterases-1 (Ppt1)-protein and Ppt1-enzyme activity in the lysosome. J Inherit Metab Dis 2019;42:944–954.
- Mukherjee AB, Appu AP, Sadhukhan T, Casey S, Mondal A, Zhang Z, Bagh MB. Emerging new roles of the lysosome and neuronal ceroid lipofuscinoses. Mol Neurodegener 2019;14(1):4.
- Sarkar C, Sadhukhan T, Bagh MB, Appu AP, Chandra G, Mondal A, Saha A, Mukherjee AB. Cln1-mutations suppress Rab7-RILP interaction and impair autophagy contributing to neuropathology in a mouse model of INCL. J Inherit Metab Dis 2020;43:1082–1101.
- Sadhukhan T, Bagh MB, Sadhukhan S, Appu AP, Mondal A, Iben JR, Li T, Coon SL, Mukherjee AB. Ablation of microRNA-155 and neuroinflammation in a mouse model of CLN1-disease. Biochem Biophys Res Commun 2021;571:137–144.
- Sadhukhan T, Bagh MB, Appu AP, Mondal A, Zhang W, Liu A, Mukherjee AB. In a mouse model of INCL reduced S-palmitoylation of cytosolic thioesterase APT1 contributes to microglia proliferation and neuroinflammation. J Inherit Metab Dis 2021;44:1051–1069.
Collaborators
- Steven L. Coon, PhD, Molecular Genetics Core, NICHD, Bethesda, MD
- Aiyi Liu, PhD, Biostatistics & Bioinformatics Branch, NICHD, Bethesda, MD
- Rafael M. Previde, PhD, Section on Cellular Signaling, NICHD, Bethesda, MD
- Stanko Stojilkovic, PhD, Section on Cellular Signaling, NICHD, Bethesda, MD
- Wadih M. Zein, MD, Ophthalmic Genetics and Visual Function Branch, NEI, Bethesda, MD
Contact
For more information, email mukherja@exchange.nih.gov or visit https://www.nichd.nih.gov/research/atNICHD/Investigators/mukherjee.