Molecular Nature and Functional Role of Dendritic Voltage-Gated Ion Channels

- Dax Hoffman, PhD, Head, Molecular Neurophysiology and Biophysics Section
- Jiahua Hu, PhD, Staff Scientist
- Lin Lin, PhD, Microbiologist
- Ying Liu, MD, Biologist
- Cole Malloy, PhD, Postdoctoral Fellow
- Jon Murphy, PhD, Postdoctoral Fellow
- Meghyn Welch, PhD, Postdoctoral Fellow
- Maisie Ahern, BS, Postbaccalaureate Fellow
- Ashley Pratt, BS, Postbaccalaureate Fellow
The central nervous system (CNS) underlies all our experiences, actions, emotions, knowledge, and memories. With billions of neurons each firing hundreds of times per second, the complexity of the brain is stunning. To pare down the task of understanding something so complex, our research approach calls for studying the workings of a single central neuron: the pyramidal neuron from the CA1 region of the hippocampus. The hippocampus is essential for long-term memory in humans and is among the first brain regions affected by epilepsy and Alzheimer’s disease. To understand how the hippocampus stores and processes information, we focus on the CA1 pyramidal neuron, one of its principal cell types. Each of these cells receives tens of thousands of inputs onto its dendrites, and it is commonly thought that information is stored by altering the strength of individual synapses (synaptic plasticity). Recent evidence suggests that the regulation of synaptic surface expression of glutamate receptors can, in part, determine synaptic strength. However, the dendrites contain an abundance of ion channels that are involved in receiving, transforming, and relaying information in the dendrites, adding an additional layer of complexity to neuronal information processing.
We found that the A-type potassium channel subunit Kv4.2 is highly expressed in the dendritic regions of CA1 neurons in the hippocampus and, as one of the primary regulators of dendritic excitability, plays a pivotal role in information processing. Kv4.2 is targeted for modulation during the types of plasticity thought to underlie learning and memory. Moreover, we found that the functional expression level of Kv4.2 regulates the subtype expression of NMDA–type glutamate receptors, the predominant molecular devices controlling synaptic plasticity and memory. We are currently following up on these findings with more detailed investigations into the mechanisms of activity-dependent Kv4.2 regulation. In addition, we have begun to investigate the role of dendritic voltage-gated potassium and calcium channels in neuronal development and developmental disorders.
Role of voltage-gated ion channels in synaptic development and disease
Isomerase regulation of potassium channel trafficking and function
The transient voltage-gated K+ current (IA), mediated by Kv4.2 in CA1 hippocampal pyramidal neurons, regulates dendritic excitability, synaptic plasticity, and learning. We recently identified a novel molecular cascade initiated by the activation of p38 kinase and subsequent Pin1–dependent isomerization of a C-terminal motif (T607) in Kv4.2 that triggers dissociation from its auxiliary subunit DPP6, a reduction IA, and an increase in neuronal excitability. Pin1 is a prolyl isomerase that selectively binds to and isomerizes phospho-Ser/Thr-Pro (pSer/Thr-Pro) bonds. Mis-regulation of Pin1 plays an important role in a growing number of pathological conditions including Alzheimer's disease, where it may protect against age-dependent neurodegeneration. We identified Pin1 as a Kv4.2–binding partner via a TAP-MS pulldown assay. Subsequent biochemical studies revealed that Pin1–Kv4.2 binding is direct and via the canonical Pin1–binding motif. Stimuli including seizure induction and exposure to enriched, novel environments increased Kv4.2 phosphorylation at the Pin1 binding site T607 by p38 MAPK in the mouse cortex and hippocampus. Using biochemical and electrophysiological techniques, we showed that Pin1 activity is required for the dissociation of the Kv4.2–DPP6 complex and that this action alters neuronal excitability. To investigate the consequences of this cascade on behavior and neuronal physiology, we used CRISPR-Cas9 techniques to generate a knockin mouse in which the isomerase binding site is specifically abolished (Kv4.2TA). The mice are viable and appear normal, although the activity-dependent dissociation of the Kv4.2–DPP6 complex is impaired.
Cole Malloy used patch-clamp electrophysiology in pyramidal cells of hippocampal slices from Kv4.2TA and wild-type (WT) mice to decipher the role of p38-Pin1–mediated regulation of Kv4.2 on neuronal excitability. He found that Kv4.2TA cells displayed lower action potential (AP) firing than in WT cells in response to somatic current injections. The reduced excitability can be traced to increased Kv4.2–mediated current in Kv4.2TA cells in outside-out somatic patches. Pharmacological block of both p38 kinase and Pin1 in WT recapitulated the impact of the mutation on neuronal firing properties and IA, confirming the specificity of the cascade underlying such effects.
To detect how these alterations in neuronal physiology may manifest in behavioral changes, Jiahua Hu performed a battery of tests probing seizure susceptibility and learning and memory capability. In response to IP kainic acid injection, Kv4.2TA mice exhibited lower seizure intensity over an hour-long period than did WT mice. The reduced seizure intensity also could be recapitulated in WT mice with pharmacological block of p38 kinase and Pin1. We have therefore identified a novel signaling cascade that can be a target for therapeutic intervention to mitigate seizure intensity in epilepsy by reducing Kv4.2 downregulation.
Kv4.2TA mice exhibit normal initial learning and memory in the Morris Water Maze. However they exhibited better ‘reversal’ learning in the Morris Water Maze than did WT mice. The data strongly support the idea that activity-dependent regulation of Kv4.2 plays an important role in cognitive flexibility. Cognitive flexibility is the ability to appropriately adjust one’s behavior to a changing environment and is impaired in various neurodevelopmental disorders, such as the autism spectrum disorder. Considering the finding that Kv4.2TA mice exhibit enhanced cognitive flexibility, ongoing experiments are investigating potential differences in synaptic properties between WT and Kv4.2TA mice. Collectively, such experiments will reveal the cellular mechanisms underlying the reversal learning phenotype in Kv4.2TA mice and will provide further insight into mechanisms impacting cognitive flexibility.
Ca2+ regulation of potassium channel function
Jonathan Murphy found that Ca2+ entry mediated by the voltage-gated Ca2+ channel subunit Cav2.3 regulates Kv4.2 function both in a heterologous expression system and endogenously in CA1 pyramidal neurons through Ca2+ binding to auxiliary subunits known as K+ channel–interacting proteins (KChIPs). KChIPs are calcium-sensing molecules containing four EF-hands, which are dysregulated in several diseases and disorders including epilepsy, Huntington’s disease, and Alzheimer’s disease. He characterized a KChIP–independent interaction between Cav2.3 and Kv4.2 using immunofluorescence colocalization, coimmunoprecipitation, electron microscopy, (FRAP (fluorescence recovery after photobleaching), and FRET (fluorescence resonance energy transfer). We found that Ca2+ entry via Cav2.3 increases Kv4.2–mediated whole-cell current, which is attributable in part to an increase in Kv4.2 surface expression. In hippocampal neurons, pharmacological block of Cav2.3 reduced whole-cell IA. We also found a reduction in whole-cell IA in Cav2.3 knockout (KO) mice mouse neurons with a loss of the characteristic dendritic IA gradient. Furthermore, the Cav2.3–Kv4.2 complex was found to regulate the size of synaptic currents and spine Ca2+ transients. The results reveal an intermolecular Cav2.3–Kv4.2 complex impacting synaptic integration in CA1 hippocampal neurons. To directly test whether the binding interaction of Cav2.3 and Kv4.2 is required for Cav2.3 regulation of Kv4.2 function, we have worked toward developing tools to disrupt the Cav2.3–Kv4.2 interaction while sparing the expression of Cav2.3.
The KChIP protein, but not mRNA expression, has been shown to be reduced in Kv4.2 KO mouse brains, suggesting increased KChIP protein degradation in the absence of Kv4.2. We hypothesized that KChIP protein degradation depends on binding to Kv4.2 and that there is increased KChIP protein degradation in the absence of Kv4.2. We aimed to elucidate the undetermined molecular mechanism of KChIP protein degradation and its effect on Kv4.2 protein levels and function. While a member of the Section, former postbaccalaureate fellow Joe Krzeski identified the pathway through which KChIP is degraded and a novel function for KChIP regulation of Kv4.2. Jiahua Hu generated a conditional Kv4.2 KO mouse using CRISPR-Cas9 techniques. Joe Krzeski injected AAV-CRE-GFP virus into the CA1 in hippocampus of the conditional Kv4.2 KO mice. We found that the Kv4.2 protein level is significantly reduced in the CRE–positive area. Interestingly, the KChIP protein level is also significantly reduced in the same area. These data suggest that KChIP protein can be dynamically regulated by Kv4.2 expression. Krzeski identified a conserved lysine residue that can be ubiquitinated. Further studies will elucidate the mechanism of KChIP degradation and its regulation by Kv4.2. A mechanistic understanding of KChIP protein degradation is important, as it may lead to new therapeutic strategies to treat diseases in which KChIPs are dysregulated.
DPP6 impacts brain development and behavior.
We previously showed that the Kv4 auxiliary subunit DPP6 has a novel function in regulating dendritic filopodia formation and stability, affecting synaptic development and function (Lin et al., Nat Commun 2013;4:2270). Recently, using immunofluorescence and electron microscopy, in a project lead by Lin Lin, we discovered a novel structure in hippocampal area CA1 that was significantly more prevalent in DPP6–KO mice than in WT mice of the same age and that such structures were observed earlier in development in DPP6–KO mice. The novel structures appeared as clusters of large puncta that colocalized NeuN, synaptophysin, and chromogranin A. Electron microscopy revealed that the structures are abnormal, enlarged presynaptic swellings filled with mainly fibrous material with occasional peripheral, presynaptic active zones forming synapses. We found diagnostic biomarkers of Alzheimer’s disease present in abnormal levels in DPP6–KO mice, including accumulation of amyloid and amyloid precursor protein (APP) in the hippocampal CA1 area and a significant increase in the expression of hyper-phosphorylated tau. The amyloid and phosphorylated tau pathologies were associated with neuroinflammation characterized by activation of microglia and astrocytes. We also found that activated astrocytes and microglia were significantly increased in DPP6–KO brain sections. We showed that DPP6–KO mice display circadian dysfunction, a common symptom of Alzheimer’s disease. Taken together, the results indicate that DPP6–KO mice show symptoms of enhanced neurodegeneration reminiscent of Alzheimer’s disease associated with a novel structure resulting from synapse loss and neuronal death. We continue to investigate DPP6 in neurodegeneration.
Figure 1. Aging DPP6–KO mice have a novel structure with characteristics of abnormal presynaptic terminals and amyloid plaques in hippocampus.
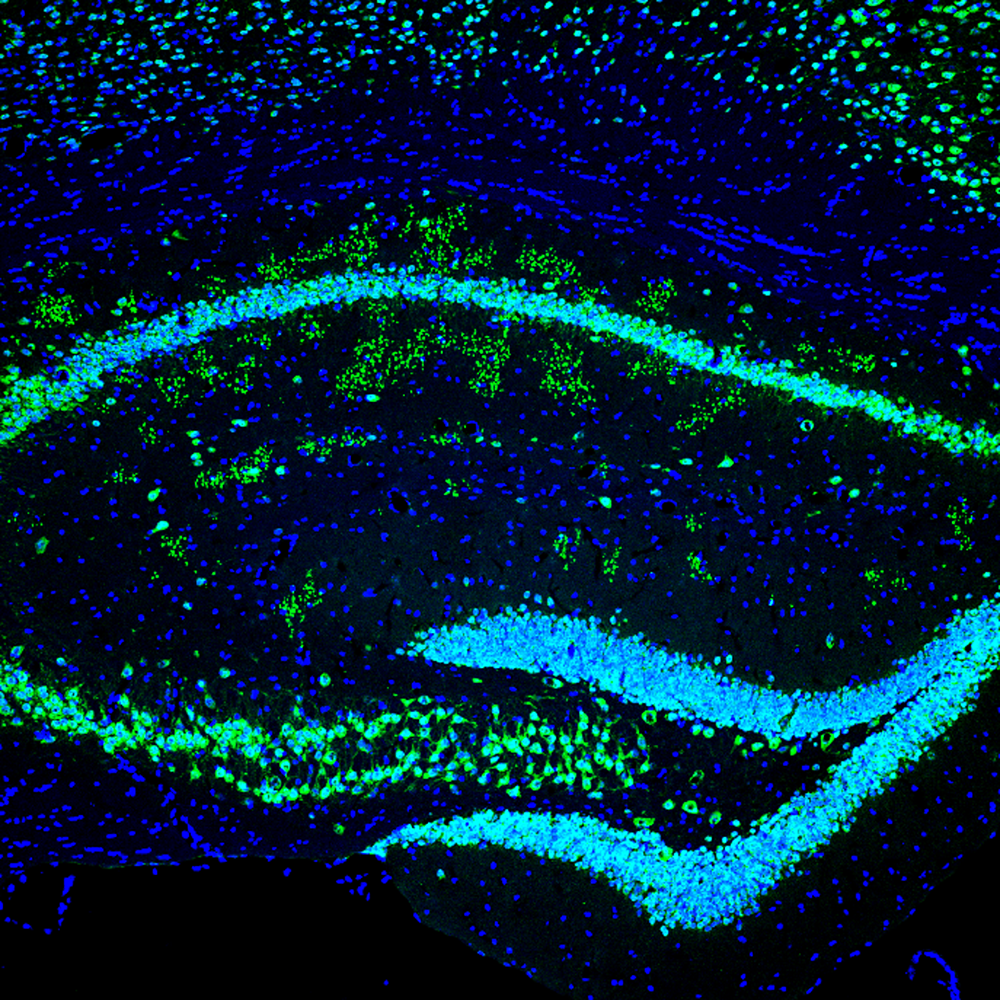
Click image to view.
Green staining NeuN, Blue DAPI
Publications
- Hu JH, Malloy C, Tabor GT, Gutzmann JJ, Liu Y, Abebe D, Karlsson RM, Durell S, Cameron HA, Hoffman DA. Activity-dependent isomerization of Kv4.2 by Pin1 regulates cognitive flexibility. Nat Commun 2020;11(1):1567.
- Hu JH, Malloy C, Hoffman DA. P38 regulates kainic acid-induced seizure and neuronal firing via Kv4.2 phosphorylation. Int J Mol Sci 2020;21(16):5921.
- Lin L, Petralia RS, Lake R, Wang YX, Hoffman DA. A novel structure associated with aging is augmented in the DPP6-KO mouse brain. Acta Neuropathol Commun 2020;8(1):197.
- Gray EE, Murphy JG, Liu Y, Trang I, Tabor GT, Lin L, Hoffman DA. Disruption of GpI mGluR-dependent Cav2.3 translation in a mouse model of fragile X syndrome. J Neurosci 2019;39(38):7453–7464.
- Murphy JG, Hoffman DA. A polybasic motif in alternatively spliced KChIP2 isoforms prevents Ca2+ regulation of Kv4 channels. J Biol Chem 2019;294(10):3683–3695.
- Gutzmann JJ, Lin L, Hoffman DA. Functional coupling of Cav2.3 and BK potassium channels regulates action potential repolarization and short-term plasticity in the mouse hippocampus. Front Cell Neurosci 2019;13:27.
Collaborators
- Heather Cameron, Section on Neuroplasticity, NIMH, Bethesda, MD
- Constantine A. Stratakis, MD, D(med)Sci, Section on Endocrinology and Genetics, NICHD, Bethesda, MD
Contact
For more information, email hoffmand@mail.nih.gov or visit https://hoffmanlab.nichd.nih.gov.