Cell Fusion Stages of Myogenesis and Osteoclastogenesis: Mechanisms and Physiological Role

- Leonid V. Chernomordik, PhD, Head, Section on Membrane Biology
- Eugenia Leikina, DVM, Senior Research Assistant
- Kamram Melikov, PhD, Staff Scientist
- Elena Zaitseva, PhD, Staff Scientist
- Jarred Whitlock, PhD, Intramural Research Training Award Fellow
- Eliana Huffman, BS, Postbaccalaureate Fellow
Diverse biological processes, in which enveloped viruses infect cells and cells from all kingdoms of life secrete, internalize, traffic and sort integral proteins, sculpt their membranes and bring together parent genomes in sexual reproduction, share a common stage: fusion of two membranes into one. Biological membrane remodeling is tightly controlled by protein machinery but is also dependent on the lipid composition of the membranes. Whereas each kind of protein has its own individual personality, membrane lipid bilayers have rather general properties, manifested by their resistance to disruption and bending and by their charge. Our long-term goal is to understand how proteins fuse membrane lipid bilayers. We expect that a better understanding of important fusion reactions will bring about new ways of controlling them and lead to new strategies for quelling diseases involving cell invasion by enveloped viruses and defects in intracellular trafficking or in intercellular fusion. Our general strategy is to combine in-depth analysis of the best characterized fusion reactions with comparative analysis of diverse, less explored fusion reactions that can reveal new kinds of fusion proteins and clarify the generality of emerging mechanistic insights. In our recent studies, we explored the mechanisms of myoblast fusion in the formation of multinucleated myofibers and the link between efficiency of formation of multinucleated osteoclasts and several IL-1–mediated diseases.
Myomerger promotes fusion pore by elastic coupling between proximal membrane leaflets and hemifusion diaphragm.
Myoblast fusion, a key process in the development and regeneration of skeletal muscle, is one of the best characterized examples of cell-cell fusion [Brukman, Uygur, Podbilewicz, Chernomordik J Cell Biol 2019;218:1436]. Two muscle-specific proteins were discovered to be essential for myoblast fusion: Myomaker [Millay et al. Nature 2013;499:301] and Myomerger/Myomixer/Minion [Bi P et al., Science 2017;356:323; Quinn et al. Nat Commun 2017;8:15665; Zhang et al. Nat Commun 2017;8:15664]. In our earlier work, we found that Myomaker is required for hemifusion, whereas the subsequent transition from hemifusion to complete fusion depends on the extracellularly localized region (ectodomain) of Myomerger [Reference 1]. The role of Myomerger in myoblast fusion represents a paradigm applicable to all protein-mediated fusion events, the essence of which is the promotion of fusion-pore formation in the hemifusion diaphragm (HD), which consists of the distal (inner) monolayers of the fusing membranes by proteins/protein regions that do not directly interact with these membrane monolayers. Considering that Myomerger is not involved in hemifusion but drives the transition from hemifusion to fusion, this protein presents an important model for analysis of the physical mechanisms underlying an, apparently, indirect effect of Myomerger and other proximal leaflet-associated factors on the fusion pore formation.
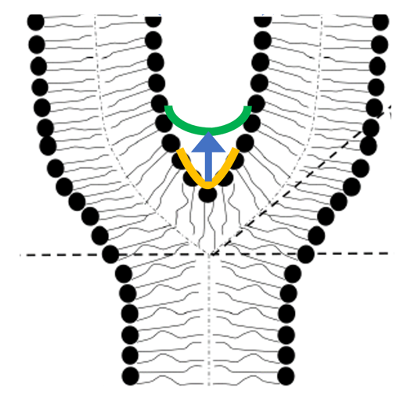
In our recent study [Reference 2], collaboration with the lab of Michael Kozlov, to explain how Myomerger promotes hemi-to-full fusion transition in myoblast fusion, we hypothesized that Myomerger shifts the spontaneous curvature of the proximal membrane monolayers towards positive values (Figure 1). The shift generates additional tension in the distal monolayers composing the HD and, hence, promotes the fusion-pore formation. We theoretically analyzed the effects of the positive spontaneous curvature of proximal membrane monolayers on the elastic stresses in HD and uncovered an HD rim–mediated elastic crosstalk between the proximal membrane monolayers and the HD. The crosstalk elicits growing HD tension and fusion pore-formation following increased proximal monolayer spontaneous curvature.
We supported the suggested mechanism by experiments in which we found that a synthetic polypeptide sMyomerger26-84, with an amino acid sequence corresponding to Myomerger protein lacking the N-terminal transmembrane domain of the protein (amino acids 1-25), generates positive spontaneous curvature of the lipid monolayer. Furthermore, the fusion defect in Myomerger-deficient myoblasts can be partially rescued not only by application of sMyomerger26-84 but also by application of lysophosphatidylcholine (LPC), a lipid of positive spontaneous curvature. A sufficiently strong shift of the spontaneous curvature in the proximal monolayers to positive values by LPC or by sMyomerger26-84 inhibited hemifusion and, consequently, fusion. However, the concentrations of sMyomerger26-84 and LPC that promoted hemifusion-to-fusion transition were considerably lower than those required for the hemifusion inhibition. We suggest that levels of Myomerger expression characteristic for fusion-committed myoblasts are in a range that allows Myomerger to promote rather than inhibit myoblast fusion. Our estimate of the surface density of sMyomerger26-84 that rescues fusion of Myomerger-deficient cells is comparable to the surface densities reported for viral fusogens. The physical mechanism, by which, according to our analysis, Myomerger drives fusion pore opening, can also underlie the effects of other proximal leaflet-associated factors on the fusion pore formation in diverse fusion processes.
Figure 1. The hypothetical mechanism for the function of Myomerger in myoblast fusion
Click image to view.
We suggest that a Myomerger-induced increase in the spontaneous curvature of the proximal monolayers of the fusing membrane bilayers changes the junction angle and the hemifusion diaphragm radius. This generates additional elastic stresses in the distal monolayers, resulting in an extra lateral tension in the diaphragm that accelerates the fusion-pore formation.
Novel Majeed syndrome–causing LPIN2 mutations link bone inflammation to inflammatory M2 macrophages and accelerated osteoclastogenesis.
In our recent work, as a part of multi-laboratory collaboration led by Raphaela Goldbach-Mansky, we identified novel heterozygous mutations in lipin 2 (LPIN2), a magnesium-dependent phosphatide phosphatase enzyme, in a patient with Majeed syndrome, and we explored the mechanisms that link these mutations to the development of sterile osteomyelitis [Reference 3]. Mutations in LPIN) differentially affect monocytes, monocyte-derived M1-like macrophages, and M2-macrophages, as well as osteoclastogenesis in various IL-1 (interleukin-1)–mediated diseases. Our data indicate that the mutations promote accelerated fusion of M2–like macrophage osteoclast precursors and the formation of multinucleated osteoclasts. The promotion of osteoclast fusion is associated with higher levels of nuclear factor–activated T cells c1 (NFATc1), a master transcription regulator of osteoclast differentiation that controls several osteoclast-specific genes, such as TRAP and cathepsin, and is required for sufficient osteoclast differentiation. LPIN2 deficiency also enhances the mitogen-activated protein kinase (MAPK) signaling pathway, including c-Jun N-terminal kinase (JNK), another important regulator of osteoclast differentiation activated by the pro-inflammatory cytokine IL-1. The proinflammatory effects of the LPIN2 mutation and fusion of osteoclasts were inhibited by IL-1 blockade with the human IL-1 receptor antagonist Anakinra. Our findings suggest a novel role of LPIN2 in macrophage polarization and the formation of bone-resorbing multinucleated osteoclasts, and they provide a model for the pathogenesis of sterile osteomyelitis, which distinguishes the Majeed syndrome from other IL-1–mediated autoinflammatory diseases.
Additional Funding
- NICHD Director’s Awards, 2020, 2021
- Office of Aids Research Award, 2019, 2020, 2021
- NICHD’s Strategic Plan 2020, extended into 2021
Publications
- Leikina E, Gamage DG, Prasad V, Goykhberg J, Crowe M, Diao J, Kozlov MM, Chernomordik LV, Millay DP. Myomaker and Myomerger work independently to control distinct steps of membrane remodeling during myoblast fusion. Dev Cell 2018;46:767-780.
- Golani G, Leikina E, Melikov K, Whitlock JM, Gamage DG, Luoma-Overstreet G, Millay DP, Kozlov MM, Chernomordik LV. Myomerger promotes fusion pore by elastic coupling between proximal membrane leaflets and hemifusion diaphragm. Nat Commun 2021;12:495.
- Bhuyan F, de Jesus AA, Mitchell J, Leikina E, VanTries R, Herzog R, Onel KB, Oler A, Montealegre Sanchez GA, Johnson KA, Bichell L, Marrero B, De Castro LF, Huang Y, Calvo KR, Collins M, Ganesan S, Chernomordik LV, Ferguson PJ, Goldbach-Mansky R. Novel Majeed syndrome-causing LPIN2 mutations link bone inflammation to inflammatory M2 macrophages and accelerated osteoclastogenesis. Arthritis Rheumatol 2021;73:1021-1032.
- Whitlock J, Chernomordik LV. Flagging fusion: phosphatidylserine signaling in cell-cell fusion. J Biol Chem 2021;296:100411.
- Whitlock J. The taming of a scramblase. J Gen Physiol 2021;153:e202012831.
Collaborators
- Michael Collins, MD, Skeletal Disorders & Mineral Homeostasis Section, NIDCR, Bethesda, MD
- Raphaela T. Goldbach-Mansky, MD, MHS, Laboratory of Clinical Immunology & Microbiology, NIAID, Bethesda, MD
- Michael M. Kozlov, PhD, Dhabil, Sackler Faculty of Medicine, Tel Aviv University, Tel Aviv, Israel
- Leonid Margolis, PhD, Section on Intercellular Interactions, NICHD, Bethesda, MD
- Richard Maraia, MD, Section on Molecular and Cellular Biology, NICHD, Bethesda, MD
- Douglas Millay, PhD, Cincinnati Children's Hospital Medical Center, Cincinnati, OH
Contact
For more information, email chernoml@mail.nih.gov or visit https://www.nichd.nih.gov/research/atNICHD/Investigators/chernomordik.