Regulation of Childhood Growth

- Jeffrey Baron, MD, Head, Section on Growth and Development
- Kevin Barnes, PhD, Senior Research Assistant
- Julian Lui, PhD, Staff Scientist
- Youn Hee Jee, MD, Staff Clinician
- Benjamin Hauser, BS, Postbaccalaureate Fellow
- Adrian Temnycky, BS, Postbaccalaureate Fellow
- Jacob Wagner, BS, Postbaccalaureate Fellow
- Elaine Zhou, BA, Postbaccalaureate Fellow
- Natalia Wojnowski, BS, Medical Research Scholars Fellow
Section on Growth and Development
Click image to view.
First row (left to right): Kevin Barnes, Youn Hee Jee, Jeff Baron
Second row (left to right): Adrian Temnycky, Julian Lui, Elaine Zhou
Not pictured: Ben Hauser, Jacob Wagner, Natalia Wojnowski
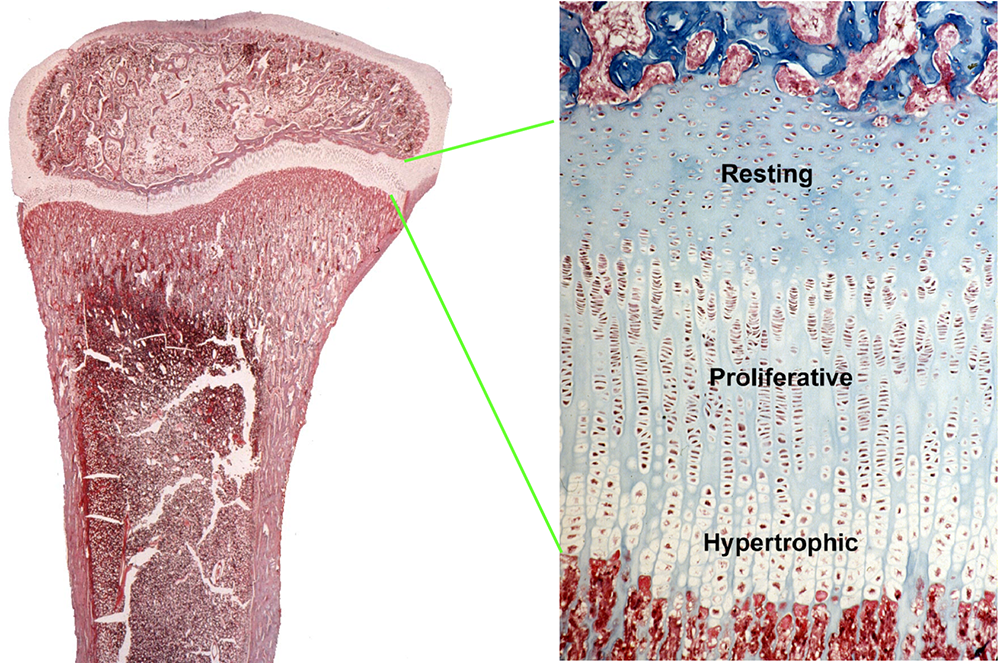
Children grow taller because their bones grow longer. Bone elongation occurs at the growth plate, a thin layer of cartilage found near the ends of juvenile bones (Figure 1). In the growth plates, new cartilage is produced through chondrocyte proliferation, hypertrophy, and cartilage matrix synthesis, and then the newly formed cartilage is remodeled into bone. The process, termed endochondral ossification, results in bone elongation, which causes children to grow in height (linear growth). Consequently, mutations in genes that regulate growth-plate chondrogenesis cause abnormal bone growth and short stature in children. Depending on the severity and nature of the genetic abnormality, the phenotype can range from chondrodysplasias with short, malformed bones, to severe, often disproportionate, short stature, to mild proportionate short stature. If the genetic defect affects tissues other than the growth-plate cartilage, the child may present with a more complex syndrome that includes other clinical abnormalities.
We investigate the cellular and molecular mechanisms governing childhood growth and development. We focus particularly on growth at the growth plate, which drives bone elongation and therefore determines height. One goal of this work is to gain insight into the many human genetic disorders that cause childhood growth failure or overgrowth. A second goal is to develop new treatments for children with severe growth disorders.
Figure 1.
Click image to view.
Histological image of a growth plate, showing the three principal zones
Novel genetic causes of childhood growth disorders
For many children who are brought to medical attention for linear growth disorders, clinical, laboratory, and genetic evaluation fails to identify the underlying etiology. Genome-wide association studies and molecular studies of growth-plate biology suggest that there are hundreds of genes that control linear growth. Therefore, there are likely many genetic causes of linear growth disorders remaining to be discovered.
To discover new genetic causes of childhood growth disorders, we invite families with monogenic growth disorders to the NIH Clinical Center, where we evaluate the clinical, biochemical, and radiological features of the condition. We then obtain DNA samples from informative family members and use powerful genetic approaches, including SNP arrays, to detect deletions, duplications, mosaicism, and uniparental disomy combined with exome sequencing to detect single-nucleotide variants and small insertions/deletions in coding regions and splice sites. When sequence variants that are likely to cause the disorder are identified, the variants and the genes in which they occur are studied in the laboratory to confirm that the variant is pathogenic, to elucidate the pathogenesis of the disorder, and to explore the role of the gene in normal growth.
We used this approach to identify new causes of childhood growth disorders. For example, we previously found that variants in aggrecan (ACAN), a component of cartilage extracellular matrix, cause autosomal-dominant short stature with advanced skeletal maturation, and that such patients also tend to develop early-onset osteoarthritis. We also found evidence that heterozygous deletion of CYP26A1 and CYP26C1, which encode enzymes that metabolize retinoic acid (RA), cause elevated RA concentrations, which accelerate bone and dental maturation in humans and cause developmental defects involving the eye and central nervous system. In additional studies, we found that variants in QRICH1, a gene of unknown function, cause a chondrodysplasia as a result of impaired growth-plate chondrocyte hypertrophic differentiation. Using this approach, we also discovered that variants in DLG2 (encoding a membrane-associated guanylate kinase) cause delayed puberty and contribute to isolated hypogondotropic hypogonadism by a mechanism that involves decreased NMDA receptor phosphorylation and signaling, and consequently decreased GnRH expression [Reference 1].
We recently used similar approaches to study the etiology of congenital hypopituitarism. Children with this disorder present with pituitary hormone deficiencies and can have a small or absent anterior pituitary gland and an ectopic posterior pituitary. Occasionally the condition is familial, but more often it occurs sporadically. In most patients, the etiology remains unknown. We studied 13 children with sporadic congenital hypopituitarism. Children with non-endocrine, non-familial idiopathic short stature served as a control group. Exome sequencing was performed in each proband and both unaffected parents. We used a burden-testing approach to compare the number of candidate variants in the two groups. First, we assessed the frequency of rare, predicted pathogenic variants in 42 genes previously reported to be associated with pituitary gland development. The average number of variants per individual was greater in probands with congenital hypopituitarism than those with non-familial short stature. Similarly, the number of probands with at least one variant in a pituitary-associated gene was greater in congenital hypopituitarism than in non-familial short stature. Second, we assessed the frequency of rare, predicted pathogenic variants in any protein-coding gene in the genome (to capture undiscovered causes) that were inherited in a fashion that could explain the sporadic occurrence of the probands’ condition (congenital hypopituitarism or non-familial short stature) with a monogenic etiology (de novo mutation, autosomal recessive, or X-linked recessive). There were fewer monogenic candidates in probands with congenital hypopituitarism than those with non-familial short stature. Our findings provide evidence that the etiology of sporadic congenital hypopituitarism has a major genetic component but is infrequently monogenic with full penetrance, suggesting that the disorder often has a more complex etiology [Reference 2].
Figure 2.
Click image to view.
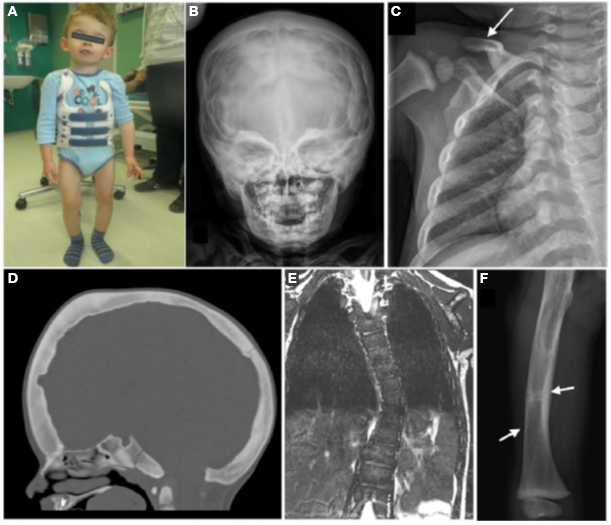
A child with a complex skeletal dysplasia caused by a neomorphic variant in SP7
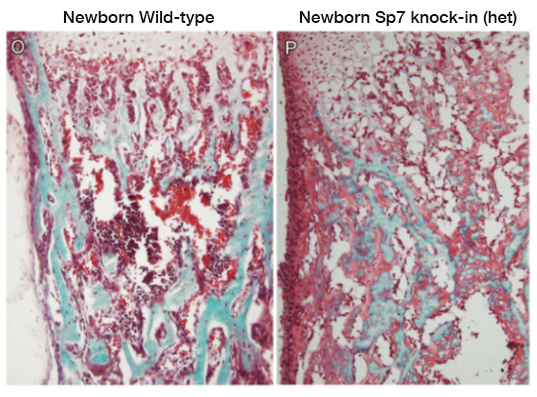
We also recently studied a child with a complex skeletal dysplasia, which included severe scoliosis, thickened calvarium, craniosynostosis, osteosclerosis of the clavicles and spine, and recurring fractures in the lower extremities (Figure 2). Some regions of the skeleton showed increased bone density and others showed decreased bone density. The number of osteoblasts was elevated but bone mineralization was impaired. We found that the disorder was caused by a de novo mutation in SP7, a gene also known as osterix. SP7 encodes a transcription factor required for differentiation of osteoblasts, a cell type required for bone formation. Previously, recessive loss-of-function sequence variants in SP7 were identified as a cause of osteogenesis imperfecta in which bone formation is markedly diminished, a phenotype strikingly different from that of the subject we studied. We generated mice with a variant orthologous to that of our subject. The mice showed a similar complex skeletal phenotype, confirming that the variant was pathogenic (Figure 3). The mutation shifted the DNA–binding specificity of SP7 from AT-rich motifs to a GC–consensus sequence (typical of other SP family members), resulting in an aberrant gene expression profile and abnormal osteoblast differentiation (Figure 4). Our study identifies a novel pathogenic mechanism in which a mutation in a transcription factor shifts DNA–binding specificity and provides the first in vivo evidence that the affinity of SP7 for AT–rich motifs, unique among SP proteins, is critical for normal osteoblast differentiation [Reference 3].
Figure 3.
Click image to view.
Bone histology from mice with an Sp7 mutation that is orthologous to the mutation found in the study subject
Figure 4.
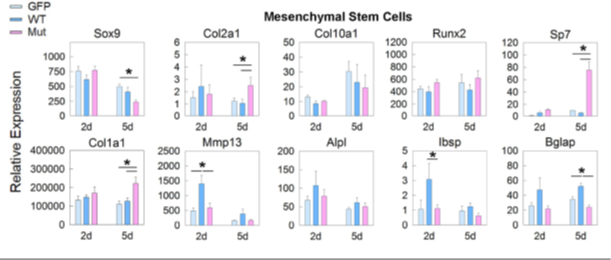
Click image to view.
The pathogenic SP7 variant produces an aberrant gene expression profile when expressed in mesenchymal stem cells.
Molecular and cellular mechanisms by which specific genes and pathways regulate childhood growth
Our group also studies the fundamental mechanisms governing skeletal growth. Much of our work has focused on the growth plate, a thin layer of cartilage which is responsible for bone growth in children and therefore for height gain. Growth at the growth plate is controlled by many interacting regulatory systems, involving endocrine, paracrine, extracellular matrix-related, and intracellular pathways. Previously, our group studied growth-plate regulation by FGFs (fibroblast growth factors), BMPs (bone morphometric proteins), C-type natriuretic peptide, retinoids, WNTs (growth factors), PTHrP/IHH (signaling pathway that regulates chondrocyte differentiation), IGFs (insulin-like growth factors), estrogens, glucocorticoids, and microRNAs. More recently, we showed evidence that SOX9, a transcription factor, regulates the trans-differentiation of growth-plate chondrocytes into osteoblasts.
We also investigated the mechanisms that cause bone growth to occur rapidly in early life but then to progressively slow with age and eventually cease. We found evidence that the developmental program responsible for the decline in growth-plate function plays out more slowly in larger bones than in smaller bones and that such differential aging contributes to the disparities in bone length and therefore to establishing normal mammalian skeletal proportions [Reference 4].
We also studied the role of EZH2 in the growth plate. EZH2 encodes a histone methyltransferase that catalyzes the trimethylation of histone H3 at lysine 27 (H3K27), which serves as an epigenetic signal for chromatin condensation and transcriptional repression. We found that loss of EZH2 and its paralog EZH1 in mice impairs bone growth, and we explored the cellular and molecular mechanisms involved. Although loss of EZH1/2 impairs growth, some heterozygous missense variants in this gene cause the Weaver overgrowth syndrome. We found that the variants responsible for the Weaver syndrome cause a partial loss of function with reduced histone methyltransferase activity. We created a mouse model that showed mild overgrowth, recapitulating the Weaver phenotype. Thus, our findings demonstrate that the Weaver syndrome is the result of EZH2 variants that cause a partial loss of function.
New treatment approaches for growth plate disorders
Recombinant human growth hormone (GH) is commonly used to treat short stature in children. However, GH treatment has limited efficacy, particularly in severe, non-GH–deficient conditions such as chondrodysplasias, and has off-target effects. Systemic insulin-like growth factor-1 (IGF-1) treatment has similar deficiencies. There are many endocrine and paracrine factors that promote chondrogenesis at the growth plate, which could potentially be used to treat such disorders. Targeting these growth factors specifically to the growth plate might augment the therapeutic skeletal effect while diminishing undesirable effects on non-target tissues. To develop growth plate–targeted therapy, we previously used yeast display to identify single-chain human antibody fragments that bind to cartilage with high affinity and specificity. As a first test of this approach, we created fusion proteins combining the cartilage-targeting antibody fragments with IGF-1, an endocrine/paracrine factor that positively regulates chondrogenesis. Such fusion proteins retained both cartilage binding and IGF-1 biological activity and were able to stimulate bone growth in an organ culture system. Using a GH–deficient mouse model, we found that subcutaneous injections of the fusion proteins increased growth-plate height without increasing proliferation in kidney cortical cells, demonstrating greater on-target efficacy at the growth plate and less off-target effect on the kidney than IGF-1 alone. Our findings provide proof of principle that targeting therapeutics to growth-plate cartilage can potentially improve treatment for childhood growth disorders [Reference 5].
We are currently applying this approach to target other chondrogenic endocrine and paracrine factors to the growth plate. We are exploring the utility of the approach both to stimulate growth-plate chondrogenesis non-specifically and also to reverse specific genetic defects in growth-plate function by modulating the abnormal molecular pathway responsible for the growth failure.
Additional Funding
- NIH U01 award: 1U01HD086838-01A1 (2017–2021, ongoing): “Genetic Diagnosis of Childhood Growth Disorders”
Publications
- Jee YH, Won S, Lui JC, Jennings M, Whalen P, Yue S, Temnycky AG, Barnes KM, Cheetham T, Boden MG, Radovick S, Quinton R, Leschek EW, Aguilera G, Yanovski JA, Seminara SB, Crowley WF, Delaney A, Roche KW, Baron J. DLG2 variants in patients with pubertal disorders. Genet Med 2020;22:1329–1337.
- Jee YH, Gangat M, Yeliosof O, Temnycky AG, Vanapruks S, Whalen P, Gourgari E, Bleach C, Yu CH, Marhsall I, Yanovski JA, Link K, Ten S, Baron J, Radovick S. Evidence that the etiology of congenital hypopituitarism has a major genetic component but is infrequently monogenic. Front Genet 2021;12:697549.
- Lui JC, Raimann A, Hojo H, Dong L, Roschger P, Kikani B, Wintergerst U, Fratzl-Zelman N, Jee YH, Haeusler G, Baron J. A neomorphic variant in the transcription factor SP7 alters sequence specificity and causes a high-turnover bone disorder. Nat Commun 2021; in press.
- Lui JC, Jee YH, Garrison P, Iben JR, Yue S, Ad M, Nguyen Q, Kikani B, Wakabayashi Y, Baron J. Differential aging of growth plate cartilage underlies differences in bone length and thus helps determine skeletal proportions. PLoS Biol 2018;16:e2005263.
- Lui JC, Colbert M, Cheung CS, Ad M, Lee A, Zhu Z, Barnes K, Dimitrov DS, Baron J. Cartilage-targeted IGF-1 treatment to promote longitudinal bone growth. Mol Ther 2019;27:673–680.
Collaborators
- Greti Aguilera, MD, Scientist Emeritus, NICHD, Bethesda, MD
- Angela Delaney Freedman, MD, St. Jude Children’s Research Hospital, Memphis, TN
- Lijin Dong, PhD, Genetic Engineering Core, NEI, Bethesda, MD
- Ellen Leschek, MD, Division of Diabetes, Endocrinology, and Metabolic Diseases, NIDDK, Bethesda, MD
- Thomas Markello, MD, PhD, Undiagnosed Diseases Program, NHGRI, Bethesda, MD
- Madhusmita Misra, MD, Harvard Medical School, Massachusetts General Hospital, Boston, MA
- Ola Nilsson, MD, PhD, Karolinska Institute, Stockholm, Sweden
- Sally Radovick, MD, Rutgers Biomedical and Health Sciences, Robert Wood Johnson Medical School, New Brunswick, NJ
- Katherine W. Roche, PhD, Receptor Biology Section, NINDS, Bethesda, MD
- Jack Yanovski, MD, PhD, Section on Growth and Obesity, NICHD, Bethesda, MD
Contact
For more information, email jeffrey.baron@nih.gov or visit https://baron.nichd.nih.gov.