Diagnosis, Localization, Pathophysiology, and Molecular Biology of Pheochromocytoma and Paraganglioma

- Karel Pacak, MD, PhD, DSc, Head, Section on Medical Neuroendocrinology
- Marianne Knue, CRNP, Nurse Practitioner
- Sara Talvacchio, BSN, Research Nurse
- Josephine Ezemobi, CRNP, Nurse Practitioner
- Tamara Prodanov, MD, Clinical Trial Database (CTDB) Coordinator
- Thanh-Truc Huynh, MS, Biologist
- Suman Goshal, PhD, Postdoctoral Visiting Fellow
- Katerina Hadrava Vanova, PhD, Postdoctoral Fellow
- Abishek Jha, MBBS, Postdoctoral Visiting Fellow
- Ondrej Uher, MSc, Predoctoral Visiting Fellow
- Kailah Charles, BS, Predoctoral Fellow
- Mayank Patel, MD, Volunteer
- Boqun Zhu, MD, Volunteer
Pheochromocytomas (PHEOs) and paragangliomas (PGLs) are rare but clinically important chromaffin-cell tumors that typically arise, respectively, from the adrenal gland and from extra-adrenal paraganglia. The clinical features and consequences of PHEO/PGL, collectively known as PPGLs, result from the overproduction and release of catecholamines (norepinephrine and epinephrine). An undetected PHEO/PGL poses a hazard to patients undergoing surgery, childbirth, or general anesthesia because of the potential for excess catecholamine secretion, which can result in significant, often catastrophic outcomes. Diagnosing and localizing a PHEO/PGL can be challenging. Plasma and urinary catecholamines, as well as their metabolites, and radio-iodinated metaiodobenzylguanidine (MIBG) scanning can yield false-positive or false-negative results in patients harboring the tumor, and computed tomography (CT) and magnetic resonance imaging (MRI) lack sufficient specificity. The molecular mechanisms by which genotypic changes predispose to the development of PHEO/PGL remain unknown, even in patients with identified mutations. Moreover, in patients with hereditary predispositions, PPGLs differ in terms of their growth, malignant potential, catecholamine phenotype, responses to standard screening tests, various imaging modalities, and therefore to different therapeutic options. We focus on developmental, molecular, genetic, epigenetic, proteomic, metabolomic, immunologic, and other types of studies to investigate the bases for a predisposition to develop PPGLs and the expression of various neurochemical phenotypes and malignant potentials, including therapeutic responses and appropriate follow-up.
Clinical and genetic aspects of pheochromocytoma and paraganglioma
Current biomarker tests for PHEOs/PGLs are technically complex or limited. We assessed the diagnostic utility of a neuroendrocrine tumor–specific 51-marker gene blood assay (NETest) in patients with PHEOs/PGLs (n = 81), including ten pediatric patients, and age-/gender-matched controls (n = 142) using a prospective case:control (1:2) analysis. We measured mRNA (qPCR), and results were scaled from 0 to 100 (upper limit of normal < 20). NETest accuracy for PHEO/PGL diagnosis was 100%. PHEO/PGL scores were 70 ± 3 vs 8.5 ± 1 in controls. Diagnostic metrics were 94% accurate, 100% sensitive, and 92% specific. Imaging correlation with 68Ga-PET-SSA was 100%. NETest levels in PHEOs (n = 26) were significantly elevated and mixed in PHEOs/PGLs (n = 5). Adrenal-derived tumors (n = 30) exhibited significantly higher scores than did extra-adrenal–derived tumors. Cluster 2 tumors exhibited significantly higher NETest levels (n = 4: 92 ± 2) than did cluster 1 tumors (n = 35: 69 ± 4). Overall regulatory-pathway analysis identified significantly higher RAS-RAF, metastatic, pluripotential, neural, and secretory gene cluster levels in PHEOs than in PGLs. Cluster 2 PHEOs/PGLs exhibited significantly higher levels of growth-factor signaling genes than did cluster 1. The PHEOs/PGLs in the pediatric cohort (n = 10) were all NETest-positive and exhibited a gene-expression profile spectrum analogous to adults. Circulating NET transcript analysis thus identifies PHEOs/PGLs with 100% efficacy and is likely to have clinical utility in the diagnosis and management of PHEO/PGL patients.
PHEOs/PGLs are tumors with frequent mutations in genes linked to the tricarboxylic acid cycle (Krebs cycle). Until now, no pathogenic variant has been found in succinyl-CoA ligase (SUCL), an enzyme that provides substrate for succinate dehydrogenase (SDH; mitochondrial complex II; CII) and a known tumor suppressor in PHEO/PGL. A cohort of 352 subjects with apparently sporadic PHEOs/PGLs underwent genetic testing using a panel of 54 genes developed at the National Institutes of Health, including the SUCLG2 subunit of SUCL. Gene deletion, succinate levels, and protein levels were assessed in tumors where possible. To confirm the possible mechanism, we used a progenitor cell line, hPheo1, derived from a human pheochromocytoma, and ablated and re-expressed SUCLG2. We found eight germline variants in the GTP–binding domain of SUCLG2 in 15 patients (15 of 352, 4.3%) with apparently sporadic PHEO/PGL. Analysis of SUCLG2–mutated tumors and SUCLG2–deficient hPheo1 cells revealed absence of SUCLG2 protein, a reduction in the level of the SDHB subunit of CII, and faulty assembly of the complex, resulting in aberrant respiration and elevated succinate accumulation. Our study suggests SUCLG2 as a novel candidate gene in the genetic landscape of PHEO/PGL. Large-scale sequencing may uncover additional cases harboring SUCLG2 variants and provide more detailed information about their prevalence and penetrance.
Figure 1. Detection of SUCLG2 variants in PHEO/PGL patients
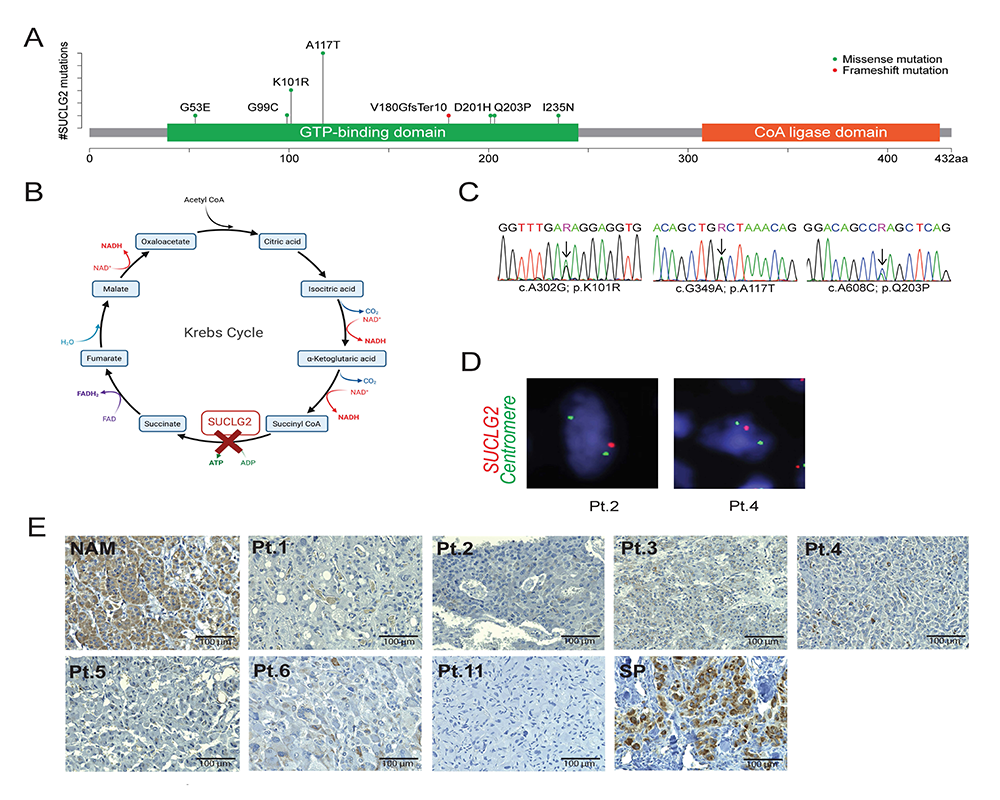
Click image to view.
A. Schematic illustration of SUCLG2 germline variants in the GTP–binding site.
B. SUCLG2 position in Krebs cycle.
C. Sanger sequencing showing SUCLG2 germline variants in patient leukocyte DNA.
D. FISH assay showing apparent heterozygous deletion of SUCLG2 in tumor specimens.
E. Immuno-histochemistry analysis revealing a substantial decrease in SUCLG2 level in PPGL with SUCLG2 germline variants compared with that in normal adrenal medulla (NAM) and sporadic PHEO/PGL (SP). Pt: patient; NAM: normal adrenal medulla; SP: sporadic PHEO/PGL.
Approximately 20% of patients diagnosed with a PHEO/PGL carry a germline mutation in one of the SDHx genes (SDHA, SDHB, SDHC, SDHD), which encode the four subunits of the SDH enzyme. When a pathogenic SDHx mutation is identified in an affected patient, genetic counseling is proposed for first-degree relatives. So far, there is no consensus on optimal initial evaluation and follow-up of individuals who are asymptomatic but might carry SDHx mutations. Thus, we established an international consensus algorithm of clinical, biochemical, and imaging screening at diagnosis and during surveillance for both adults and children. An international panel of 29 experts from 12 countries was assembled, and the Delphi method (which consists of several rounds of written questionnaires that allow experts to give their opinions) was used to reach a consensus on 41 statements. The Consensus Statement covers a range of topics, including age of first genetic testing, appropriate biochemical and imaging tests for initial tumor screening and follow-up, screening for rare SDHx–related tumors, and management of elderly people who have an SDHx mutation. This Consensus Statement focuses on the management of asymptomatic SDHx mutation carriers and provides clinicians with much needed guidance. The standardization of practice will facilitate prospective studies in the near future.
Imaging of pheochromocytomas and paragangliomas
Targeted radionuclide therapies (TRT) using 131I-metaiodobenzylguanidine (131I-MIBG) and peptide-receptor radionuclide therapy (177Lu or 90Y) represent several of the therapeutic options in the management of metastatic/inoperable pheochromocytoma/paraganglioma. Recently, high-specific-activity 131I-MIBG therapy was approved by the FDA, and both 177Lu-DOTATATE and 131I-MIBG therapy were recommended by the National Comprehensive Cancer Network guidelines for the treatment of metastatic pheochromocytoma/paraganglioma. However, a clinical dilemma often arises in the selection of TRT, especially when a patient can be treated with either type of therapy based on eligibility for MIBG and somatostatin receptor imaging. To address this problem, we assembled a group of international experts, including oncologists, endocrinologists, and nuclear-medicine physicians with substantial experience in treating neuroendocrine tumors with TRTs, to develop consensus and provide expert recommendations and perspectives on how to decide between these two therapeutic options for metastatic/inoperable pheochromocytoma/paraganglioma. Reference 3 summarizes the survival outcomes of the available TRTs, discusses personalized treatment strategies based on functional imaging scans, addresses practical issues, including regulatory approvals, and compares toxicities and risk factors across treatments. Furthermore, it discusses the emerging TRTs.
Recent professional society guidelines for radionuclide imaging of sporadic pheochromocytoma (PHEO) recommend 18F-FDOPA as the radiotracer of choice, deeming 68Ga-DOTATATE and FDG to be second- and third-line agents, respectively. An additional agent, 18F-FDA, remains experimental for PHEO detection. A paucity of research has performed head-to-head comparison among these agents. Therefore, we performed an intra-individual comparison of 68Ga-DOTATATE PET/CT, FDG PET/CT, 18F-FDOPA PET/CT, 18F-FDA PET/CT, CT, and MRI in visualization of sporadic primary PHEO. The prospective study enrolled patients referred with clinical suspicion for sporadic PHEO. Patients were scheduled for 68Ga-DOTATATE PET/CT, FDG PET/CT, 18F-FDOPA PET/CT, 18F-FDA PET/CT, whole-body staging CT (portal venous phase), and MRI, within a three-month period. Analysis included only patients with histologically confirmed PHEO on resection. We included 14 patients (8 women, 6 men; mean age, 52.4±16.8 years) with PHEO. Both 68Ga-DOTATATE PET/CT and FDG PET/CT were completed in all 14 patients, 18F-FDOPA PET/CT in 11/14, 18F-FDA PET/CT in 7 of 14, CT in 12 of 14, and MRI in 12 of 14 patients. Findings from this small intra-individual comparative study support 18F-FDOPA PET/CT as a preferred first-line imaging modality in evaluation of sporadic PHEO. In summary, the study provides data supporting current guidelines for imaging evaluation of suspected PHEO.
The aim of the next study was to identify quantitative MR biomarkers in head and neck PGLs (HNPGLs). Sixty HNPGLs were included from 50 patients. The control group consisted of 30 parapharyngeal space lesions (27 patients), which included nerve sheath tumors (n = 12) and metastatic lymph nodes (n = 18) from squamous cell carcinomas. The study identified a multi-parametric signature for disease subtyping, providing a strong impetus for switching from qualitative to quantitative analysis of deep soft-tissue tumors of the neck.
Immune and metabolic aspects of pheochromocytoma and paraganglioma
Immunotherapy has become an essential component in cancer treatment. Discovery of such tumor-specific epitopes through tumor sequencing has revolutionized patient outcomes in many types of cancers that were previously untreatable. However, the majority of solid metastatic cancers, such as PHEO, are resistant to this approach. Therefore, understanding immune cell composition in primary and distant metastatic tumors is important for therapeutic intervention and diagnostics. Combined mannan-BAM, TLR ligand, and anti-CD40 antibody-based intra-tumoral immunotherapy (MBTA therapy) previously resulted in the complete eradication of murine subcutaneous PHEO and demonstrated a systemic antitumor immune response in a metastatic model. We further evaluated this systemic effect using a bilateral PHEO model, performing MBTA therapy through injection into the primary tumor and using distant (non-injected) tumors to monitor size changes and detailed immune cell infiltration. MBTA therapy suppressed the growth not only of injected but also of distal tumors and prolonged the survival of MBTA–treated mice. Our flow-cytometry analysis showed that MBTA therapy led to increased recruitment of innate and adaptive immune cells in both tumors and the spleen. Moreover, adoptive CD4+ T cell transfer from successfully MBTA–treated mice (i.e., subcutaneous PHEO) demonstrates the importance of such cells in long-term immunological memory. In summary, the study unravels further details on the systemic effect of MBTA therapy and its use for tumor and metastasis reduction or even elimination
We further extended the MBTA therapy to other tumors and applications in close collaboration with NCI investigators. For example, emerging evidence is demonstrating the extent of T cell infiltration within the tumor micro-environment and thus has favorable prognostic and therapeutic implications. Hence, immuno-therapeutic strategies that augment the T cell signature of tumors hold promising therapeutic potential. Recently, immuno-therapy based on intra-tumoral injection of MBTA demonstrated promising potential to modulate the immune phenotype of injected tumors, including PHEO. The strategy promotes the phagocytosis of tumor cells to facilitate the recognition of tumor antigens and induce a tumor-specific adaptive immune response. Using a syngeneic colon carcinoma model, we demonstrated MBTA's potential to augment CD8+ T cell tumor infiltrate when administered intra-tumorally or subcutaneously as part of a whole tumor cell vaccine. Both immuno-therapeutic strategies proved effective in controlling tumor growth, prolonged survival, and induced immunological memory against the parental cell line. Collectively, our investigation demonstrates MBTA's potential to trigger a potent anti-tumor immune response. We also reviewed the most promising glioblastoma vaccination strategies to contextualize the MBTA vaccine. By reviewing current evidence using translational tumor models supporting MBTA vaccination, we evaluated the underlying principles that validate its clinical applicability. We also showed the translational potential of MBTA vaccination as a potential immunotherapy in glioblastoma, along with established surgical and immunologic cancer treatment paradigms.
Therapeutic aspects of pheochromocytoma and paraganglioma
PHEOs/PGLs are characterized by a unique molecular landscape that allows their assignment to clusters depending on underlying genetic alterations. With around 30–35% of Caucasian patients (a lower percentage in the Chinese population) showing germline mutations in susceptibility genes, PHEOs/PGLs have the highest rate of heritability among all tumors. A further 35–40% of Caucasian patients (a higher percentage in the Chinese population) are affected by somatic driver mutations. Thus, around 70% of all patients with PHEOs/PGLs can be assigned to one of three main molecular clusters with different phenotypes and clinical behavior. Krebs cycle/VHL/EPAS1–related cluster 1 tumors tend to have a noradrenergic biochemical phenotype and require very close follow-up because of the risk of metastasis and recurrence. In contrast, kinase signaling–related cluster 2 tumors are characterized by an adrenergic phenotype and episodic symptoms, with a generally less aggressive course. The clinical correlates of patients with Wnt signaling–related cluster 3 tumors are currently poorly described, but aggressive behavior appears likely. In a review [Reference 2], we explored and explained why cluster-specific (personalized) management of PHEO/PGL is essential to ascertain clinical behavior and prognosis and guide individual diagnostic procedures (biochemical interpretation, choice of the most sensitive imaging modalities), and personalized management and follow-up. Although cluster-specific therapy of inoperable/metastatic disease has not yet entered routine clinical practice, we suggest that informed personalized genetic-driven treatment should be implemented as a logical next step. The review published guidelines and expert views within each cluster for a coherent individualized patient management plan.
The risk factors for severe COVID-19 are diverse, yet closely resemble the clinical manifestations of catecholamine excess states (e.g., hypertension, cardiovascular disease, immune dysregulation, and hyperglycemia), suggesting a possible common basis for disease. Unfortunately, severe illness (e.g., respiratory failure, compromised cardiac function, and shock) incurred by COVID-19 hinders the direct study of catecholamines in these patients, especially among those on multiple medications or those on adrenaline or noradrenaline infusions, or both. PHEOs/PGLs are tumors that secrete catecholamines, namely adrenaline and noradrenaline, often in excess. PHEOs/PGLs are well studied disease processes in which the effects of catecholamines are easily discernible, and therefore their potential biochemical and physiological influences in patients with COVID-19 can be explored. Because catecholamines are expected to have a role in patients with critical illness, patients on vasopressor infusions, and patients who sustain some acute and chronic physical stresses, the challenges involved in the management of catecholamine excess states are directly relevant to the treatment of patients with COVID-19. In a Personal View article type, we discussed the complex interplay between catecholamines and COVID-19, and the management of catecholamine excess states, while referencing relevant insights derived from the study of PHEO/PGL.
Germline variants in approximately 20 PHEO/PGL susceptibility genes are found in about 40% of patients, half of which are found in the genes that encode SDH. Patients with SDH subunit B (SDHB)–mutated PHEO/PGL exhibit a higher likelihood of developing metastatic disease, which can be partially explained by the metabolic cell reprogramming and redox imbalance caused by the mutation. Reactive oxygen species (ROS) are highly reactive molecules involved in many important signaling pathways. A moderate level of ROS production can help regulate cellular physiology; however, an excessive level of oxidative stress can lead to tumorigenic processes, including stimulation of growth factor–dependent pathways and the induction of genetic instability. Tumor cells effectively exploit antioxidant enzymes in order to protect themselves against harmful intracellular ROS accumulation, which highlights the essential balance between ROS production and scavenging. Exploiting ROS accumulation can be used as a possible therapeutic strategy in ROS–scavenging tumor cells. We focused on the role of ROS production in PHEO and PGL, predominantly in SDHB–mutated cases. We discussed potential strategies and approaches to anticancer therapies by enhancing ROS production in these difficult-to-treat tumors.
Animal model of pheochromocytoma and cell culture studies
We previously identified the syndrome of multiple paragangliomas and pheochromocytomas, duodenal somatostatinoma, and polycythemia resulting from post-zygotic EPAS1 (HIF2A)-gain-of-function mutations (also called Pacak-Zhuang syndrome). The mutations, located in the oxygen-degradation domain (ODD) of hypoxia-inducible factor-2a (HIF-2a), have been shown to impair hydroxylation by prolyl hydroxylase domain–containing protein 2 (PHD2) and subsequent association with the von Hippel-Lindau (VHL) protein. In such a situation, degradation of HIF-2a is impaired, resulting in its stabilization, prolonged activation, lack of response to normal or increasing oxygen tension, and activation of the transcription of many genes participating in tumorigenesis. Recently, in collaboration with NCI investigators, we developed transgenic mice with a gain-of-function Epas1A529V mutation (corresponding to human EPAS1A530V), which demonstrated elevated levels of erythropoietin and polycythemia, a reduced urinary metanephrine-to-normetanephrine ratio, and increased expression of somatostatin in the ampullary region of duodenum. The findings demonstrate the vital roles of EPAS1 mutations in development of the syndrome and the great potential of the Epas1A529V animal model for further pathogenesis and therapeutics studies. The model is also being used to study other malformations in animals as well as to match them with those seen in our patients (neurological, vascular, and ocular malformations) as described below.
Patients referred to the NIH for new, recurrent, and/or metastatic PHEO/PGL were confirmed for the EPAS1 gain-of-function mutation; imaging was evaluated for vascular malformations. We evaluated the Epas1A529V transgenic syndrome mouse model, corresponding to the mutation initially detected in the patients (EPAS1A530V), for vascular malformations via intravital 2-photon microscopy of meningeal vessels, terminal vascular perfusion with Microfil silicate polymer and subsequent intact ex vivo 14T MRI and micro-CT, and histologic sectioning and staining of the brain and identified pathologies. Further, we evaluated retinas from corresponding developmental time points (P7, P14, and P21) and the adult dura via immunofluorescent labeling of vessels and confocal imaging. We identified a spectrum of vascular malformations in all nine syndromic patients and in all our tested mutant mice. Patient vessels had higher variant allele frequency than adjacent normal tissue. Veins of the murine retina and intracranial dura failed to regress normally at the expected developmental time points. The findings add vascular malformation as a new clinical feature of the EPAS1 gain-of-function syndrome.
Currently, the precise role of HIF2α in the predisposition to metastatic disease remains unclear. To assess such a role, we combined gene manipulations in PHEO cell lines with retrospective analyses of patient data and gene-expression profiling in tumor specimens. Among 425 patients with PPGLs identified with mutations in tumor-susceptibility genes, those with tumors resulting from activation of pseudohypoxic pathways had a significantly higher frequency of metastatic disease than those with tumors resulting from activation of kinase-signaling pathways, even without inclusion of patients with mutations in SDHB. Three out of nine (33%) patients with gain-of-function mutations in HIF2α had metastatic disease. In cell-line studies, elevated expression of HIF2α enhanced cell proliferation and led to increased migration and invasion capacity. Moreover, HIF2α expression in HIF2α–deficient cells resulted in increased cell motility, diffuse cluster formation, and emergence of pseudopodia, indicating changes in cell adhesion and cytoskeletal remodeling. In a mouse liver–metastasis model, Hif2a enhanced the metastatic load. Transcriptomics data revealed alterations in focal adhesion and extracellular matrix–receptor interactions in HIF2α–mutated PHEOs/PGLs. Our translational findings demonstrate that HIF2α supports pro-metastatic behavior in PHEOs/PGLs, although other factors remain critical for subsequent transition to metastasis. We identified LAMB1 (which encodes laminin subunit beta 1, an extracellular matrix glycoprotein) and COL4A2 (which encodes six subunits of type IV collagen) as new potential therapeutic targets for HIF2α–driven PHEOs/PGLs. Identified HIF2α downstream targets might open a new therapeutic window for aggressive HIF2α–expressing tumors.
Publications
- Hadrava Vanova K, Pang Y, Krobova J, Kraus M, Nahacka Z, Boukalova S, Pacak S, Zobalova R, Zhu J, Huynh T, Jochmanova I, Uher O, Garrett T, Ghayee H, Schuster B, Kanpp P, Frysack Z, Hartmann I, Nilubol N, Cerny J, Taieb D, Rohlena J, Neuzil J, Yang C, Pacak K. Germline SUCLG2 variants in patients with pheochromocytoma and paraganglioma. J Natl Cancer Inst 2022; 114(1):130–138.
- Notling S, Bechmann N, Taieb D, Beuschlein F, Fassnacht M, Kroiss M, Eisenhofer G, Grossman A, Pacak K. Personalized management of pheochromocytoma and paraganglioma. Endocr Rev 2021;27:2989.
- Jha A, Taieb D, Carrasquillo J, Pryma D, Patel M, Millo C, De Herder W, Del Rivero J, Crona J, Shulkin B, Virgolini I, Chen A, Mittal B, Basu S, Dillon J, Hope T, Aparici C, Iagaru A, Hicks R, Avaram A, Strosberg J, Civelek A, Lin F, Pandit-Tasker N, Pacak K. High-specific-activity-131I-MIBG versus 177Lu-DOTATATE targeted radionuclide therapy for metastatic pheochromocytoma and paraganglioma. Clin Cancer Res 2021;27:2989.
- Amar L, Pacak K, Steichen O, Akker S, Aylwin S, Baudin E, Buffer A, Burnichon N, Clifton-Bligh R, Dahia P, Fassnacht M, Grossman A, Herman P, Hicks R, Januszewicz A, Jimenez C, Kunst H, Lewis D, Mannelli M, Naruse M, Robledo M, Taieb D, Taylor D, Timmers H, Treglia G, Tufton N, Young W, Lenders J, Gimene-Roquelo A-P, Lussey-Lepoutre, C. International consensus on initial screening and follow-up of asymptomatic SDHx mutation carriers. Nat Rev Endcrinol 2021;17:435.
- Rosenblum J, Wang H, Dmitriev P, Cappadona A, Mastorakos P, Xu C, Jha A, Edwards N, Donahua D, Nazari MA, Knutsen R, Smirniotoloulos J, Pappo A, Spetzler R, Vortmeyer A, Gilbert M, McGavern D, Chew E, Kozel B, Heiss J, Zhuang Z, Pacak K. Developmental vascular malformations in EPAS1 gain-of-function syndrome. JCI Insight 2021;6:e144368.
Collaborators
- James Bibb, PhD, University of Alabama Comprehensive Cancer Center, University of Alabama at Birmingham Medical Center, Birmingham, AL
- Clara C. Chen, MD, Nuclear Medicine Department, Clinical Center, NIH, Bethesda, MD
- Peter Deen, PhD, Radboud Institute for Molecular Life Sciences, Nijmegen, The Netherlands
- Jaydira Del Rivero, MD, Pediatric Oncology Branch, NCI, Bethesda, MD
- Graeme Eisenhofer, PhD, Universität Dresden, Dresden, Germany
- Stephanie Fliedner, PhD, Universitätsklinikum Schleswig-Holstein, Lübeck Medizinische Klinik I, Lübeck, Germany
- Zdenek Fryšák, MD, PhD, University Hospital and Faculty of Medicine and Dentistry, Palacký University Olomouc, Olomouc, Czech Republic
- Hans Ghayee, DO, Department of Internal Medicine, University of Florida, Gainesville, FL
- Peter Herscovitch, MD, PET Department, Clinical Center, NIH, Bethesda, MD
- Frank I. Lin, MD, Molecular Imaging Program, NCI, Bethesda, MD
- W. Marston Linehan, MD, Urologic Oncology Branch, NCI, Bethesda, MD
- Corina Millo, MD, PET Department, Clinical Center, NIH, Bethesda, MD
- Jirí Neužil, PhD, Institute of Biotechnology, Czech Academy of Sciences, Prague, Czech Republic
- Seyedehmoozhan Nikpanah, MD, Nuclear Medicine Department, Clinical Center, NIH, Bethesda, MD
- Naris Nilubol, MD, FACS, Surgical Oncology Program, NCI, Bethesda, MD
- Ondrej Petrak, MD, PhD, Third Department of Medicine, General University Hospital, Prague, Czech Republic
- Margarita Raygada, PhD, Section on Endocrinology and Genetics, NICHD, Bethesda, MD
- Mercedes Robledo, PhD, Human Cancer Genetics Programme, Spanish National Cancer Centre (CNIO), Madrid, Spain
- Jared Rosenblum, MD, Neuro-Oncology Branch, NCI, Bethesda, MD
- Douglas Rosing, MD, Translational Medicine Branch, NHLBI, Bethesda, MD
- Kelly Roszko, MD, PhD, Skeletal Disorders and Mineral Homeostasis Section, NIDCR, Bethesda, MD
- Babak Saboury, MD, Nuclear Medicine Department, CC, NIH, Bethesda, MD
- Constantine A. Stratakis, MD, D(med)Sci, Section on Endocrinology and Genetics, NICHD, Bethesda, MD
- Arthur S. Tischler, MD, PhD, New England Medical Center, Boston, MA
- Richard Tothill, PhD, University of Melbourne Centre for Cancer Research, Melbourne, Australia
- Brad Wood, MD, PhD, Radiology Department, Clinical Center, NIH, Bethesda, MD
- Chunzhang Yang, PhD, Neuro-Oncology Branch, NCI, Bethesda, MD
- Deena Zeltser, MD, Office of the Clinical Director, NICHD, Bethesda, MD
- Zhengping Zhuang, MD, PhD, Neuro-Oncology Branch, NCI, Bethesda, MD
Contact
For more information, email karel@mail.nih.gov or visit https://irp.nih.gov/pi/karel-pacak.