Molecular Genetics of Heritable Human Disorders

- Janice Y. Chou, PhD, Head, Section on Cellular Differentiation
- Lisa Zhang, PhD, Staff Scientist
- Irina Arnaoutova, PhD, Staff Scientist
- Sudeep Gautam, PhD, Visiting Fellow
- Nelson George, PhD, Visiting Fellow
- Javier Anduaga, BS, Pre-Intramural Research Training Award Fellow
- Hung-Dar Chen, PhD, Contract Technician
- Cheol Lee, PhD, Contract Technician
We conduct research to delineate the pathophysiology of and develop novel therapies for the two major type I glycogen storage disease (GSD-I) subtypes, GSD-Ia and GSD-Ib. GSD-Ia is caused by a deficiency in the liver/kidney/intestine-restricted glucose-6-phosphatase-α (G6Pase-α or G6PC) and GSD-Ib is caused by a deficiency in the ubiquitously expressed glucose-6-phosphate transporter (G6PT or SLC37A4). G6Pase-α is an endoplasmic reticulum (ER)–transmembrane protein that regulates intracellular glucose production by catalyzing the hydrolysis of G6P to glucose and phosphate. The active site of G6Pase-α faces into the ER lumen and depends on G6PT, another ER transmembrane protein, and translocates G6P from the cytoplasm into the lumen. To function, G6Pase-α must couple with G6PT to form a functional G6Pase-α/G6PT complex, which maintains interprandial glucose homeostasis. GSD-Ia and GSD-Ib patients manifest a common metabolic phenotype of impaired glucose homeostasis and severe long-term complications of hepatocellular adenoma/carcinoma (HCA/HCC) and renal disease. There is no cure for GSD-Ia and GSD-Ib. We generated animal models of both disorders, which are being exploited to both delineate the disease more precisely and develop new treatment approaches, including gene therapy.
We also generated several efficacious G6Pase-α– and G6PT–expressing recombinant adeno-associated virus (rAAV) vectors and provided a proof-of-principle gene therapy in murine GSD-Ia and GSD-Ib that is safe, effective, and appropriate for clinical trials. In 2018, our GSD-Ia rAAV vector technology was licensed to Ultragenyx Pharmaceutical Inc. (Novato, CA), which is currently undertaking a phase I/II clinical trial for GSD-Ia. We have begun to explore alternative genetic technologies for GSD-I therapies. We have established formal collaborations under cooperative research and development agreements (CRADAs) with CRISPR Therapeutics (Cambridge, MA) and Beam Therapeutics (Cambridge, MA) to evaluate the efficacy of CRISPR/Cas9–based and adenine base editor (ABE)–based gene editing systems, respectively, to correct gene-specific G6PC mutations in animal models of GSD-Ia.
Correction of metabolic abnormalities in a mouse model of glycogen storage disease type Ia by CRISPR/Cas9–based gene editing
The rAAV-co-G6PC vector used in the current phase I/II clinical trial is episomally expressed, and its long-term durability of expression in humans is currently being established. We therefore sought to explore the use of the CRISPR/Cas9 technology to correct a pathogenic GSD-Ia variant in its native genetic locus. The most prevalent pathogenic mutation identified in Caucasian GSD-Ia patients is G6PC-p.R83C, representing 32% of diseased alleles. Using the CRISPR/Cas9–based gene editing technology, we generated a GSD-Ia mouse disease model, the G6pc-R83C mouse, which is homozygous for the G6PC-p.R83C mutation, and we showed that these mice manifest impaired glucose homeostasis mimicking that of human GSD-Ia. We then used a CRISPR/Cas9–based gene editing system to treat newborn G6pc-R83C mice and showed that the treated mice grew normally to age 16 weeks without hypoglycemia seizures. The treated G6pc-R83C mice, expressing 3% or more of normal hepatic G6Pase-α activity, maintained glucose homeostasis, displayed normalized blood metabolites, and could sustain 24 hours of fasting. Taken together, we have developed a second-generation therapy in which in vivo correction of a pathogenic G6PC-p.R83C variant in its native genetic locus could lead to potentially permanent, durable, long-term correction of the GSD-Ia disorder [Reference 1].
Figure 1.
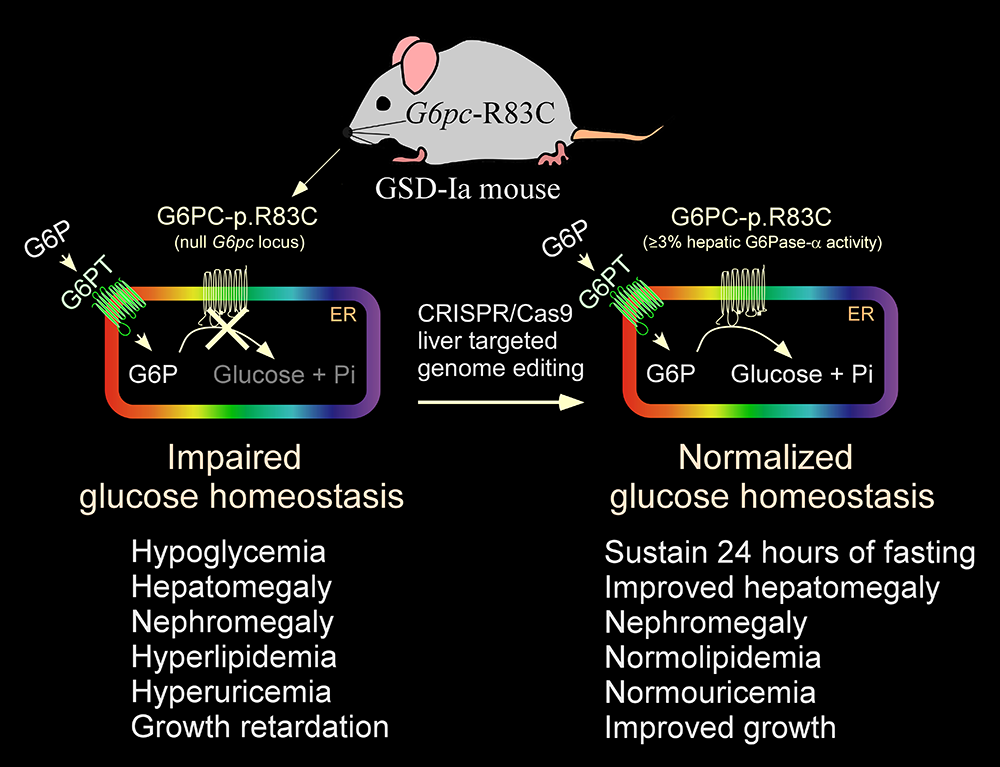
Click image to view.
Correction of metabolic abnormalities of the GSD-Ia mice by CRISPR/Cas9–based gene editing
Gene therapy for GSD-Ia
We generated four efficacious G6PC gene transfer rAAV vectors for GSD-Ia gene therapy: rAAV-G6PC that expresses the wild-type G6PC, rAAV-co-G6PC that expresses a codon-optimized (co) G6PC, rAAV-G6PC-S298C that expresses a G6PC-S298C variant with increased efficacy, and rAAV-co-G6PC-S298C. Using the rAAV-G6PC and rAAV-co-G6PC vectors, we showed that rAAV–treated G6pc–/– mice that express 3% or more of normal hepatic G6Pase-α activity maintain glucose homeostasis and show no evidence of HCA/HCC. Our GSD-Ia rAAV vector technology was commercially licensed to Ultragenyx Pharmaceutical, which has launched a phase I/II clinical trial for GSD-Ia using the rAAV-co-G6PC vector. The initial dose-escalation trial is currently in progress and no safety issues have been reported to date.
The rAAV-co-G6PC vector contains a 20% change in the native G6PC coding sequence. While routinely used in clinical therapies, codon-optimized vectors may not always be optimal. To develop alternative approaches to increase the potency of the G6PC gene-transfer vector, we generated the rAAV-G6PC-S298C and rAAV-co-G6PC-S298C vectors; the latter combines codon optimization with the S298C variant in the same construct. In a short-term (4–week) study using G6pc–/– mice, we showed that the efficacy of the rAAV-G6PC-S298C and rAAV-co-G6PC-S298C vectors were 3– and 5–fold higher than that of the rAAV-G6PC vector. We then undertook a 65–76–week study to examine the longer-term efficacy of these vectors. We infused four separate groups of G6pc–/– mice, each with one of the rAAV vectors and examined phenotypic correction of the mice at age 65–76 weeks. We showed that hepatic G6Pase-α activities in rAAV-co-G6PC–, rAAV-G6PC-S298C–, and rAAV-co-G6PC-S298C–treated G6pc–/– mice were 2.2–, 2.3–, and 6.2–fold higher, respectively, than that in rAAV-G6PC–treated mice. The rAAV-G6PC-S298C vector, which contains a 2% change in the native G6PC coding sequence, provides equal or greater efficacy to the codon optimization approach, offering a valuable alternative vector for clinical translation in human GSD-Ia. The rAAV-co-G6PC-S298C vector is three times as efficient as the rAAV-co-G6PC vector currently used in the clinical trials and thus offers a vector of choice for clinical translation in human GSD-Ia [References 2, 3].
Evaluation of the adenine base editor–based gene-editing system to correct a pathogenic G6PC mutation in a humanized mouse model of GSD-Ia
We are exploring the adenine base editor (ABE)–based technologies, which enable a programmable conversion of A•T to G•C in genomic DNA for GSD-Ia therapies. The ABE system works in both dividing and non-dividing cells, is reported to produce virtually no indels or off-target editing in the genome, can correct a pathogenic variant in its native genetic locus, and can lead to permanent, therapeutically effective long-term expression. This is a collaborative study with Beam Therapeutics, Cambridge, MA, under a CRADA.
As mentioned above, the G6PC-p.R83C is the most prevalent pathogenic mutation identified in Caucasian GSD-Ia patients. It contains a single G→A transition in the G6PC gene. We first generated a homozygous humanized (h) R83C/R83C (hR83C) mouse strain by inserting the entire coding sequence of the human G6PC-p.R83C cDNA along with human G6PC 3′-UTR into exon 1 of the mouse G6pc gene at the ATG start codon. This insertion places the human cDNA under the control of the native mouse G6pc promoter/enhancer. The mouse G6pc gene is disrupted by a premature STOP codon created in the mouse G6pc exon 1. We first showed that hR83C mice manifest impaired glucose homeostasis characterized by growth retardation, hypoglycemia, hyperlipidemia, hyperuricemia, hepatomegaly, and nephromegaly, mimicking the abnormal metabolic phenotype of human GSD-Ia. We then treated newborn hR83C mice with lipid nanoparticles encompassing the guide RNA and mRNA encoding ABE (LNP-ABE) and showed that the treated mice grew normally to age 8 weeks without hypoglycemia seizures. The LNP-ABE–treated hR83C mice expressed significant levels of hepatic G6Pase-α activity with an editing efficiency up to about 60% and displayed normalized blood metabolite profiles. Taken together, our data demonstrate the potential of base editing to correct the G6PC-p.R83C mutation and the abnormal GSD-Ia phenotype.
Molecular mechanism underlying hepatic autophagy impairment in GSD-Ib
As stated above, GSD-Ia and GSD-Ib patients manifest a common phenotype of impaired glucose homeostasis and long-term risk of HCA/HCC. The etiology of HCA/HCC in GSD-I is unknown. Autophagy is an evolutionarily conserved, degradative process that produces energy and building blocks through lysosomal degradation of intracellular proteins and organelles in times of nutrient deprivation and environmental stress. Studies have shown that a deficiency in hepatic autophagy can contribute to hepatocarcinogenesis. Autophagy can be regulated positively by sirtuin 1 (SIRT1), AMP–activated protein kinase (AMPK), and forkhead box O (FoxO) transcription factor family members. In the liver, AMPK is activated via phosphorylation of the AMPK α-subunit at residue T172 by the liver kinase B-1 (LKB1), a serine/threonine kinase which is also a tumor suppressor. We showed that G6Pase-α deficiency impairs hepatic autophagy through downregulation of SIRT1/FoxO3a signaling. We hypothesized that, in GSD-Ib, G6PT deficiency leads to hepatic autophagy deficiency.
Using mouse models of GSD-Ib, we showed that G6PT deficiency leads to impaired hepatic autophagy, evident from attenuated expression of many components of the autophagy network, impaired autophagosome formation, and reduced autophagy flux. The G6PT–deficient liver displayed impaired SIRT1 and AMPK signaling, along with reduced expression of SIRT1, FoxO3a, LKB1, and p-AMPK. Hepatic overexpression of either SIRT1 or LKB1 in the G6PT–deficient liver restored autophagy and SIRT1/FoxO3a/LKB1/AMPK signaling. The hepatosteatosis in GSD-Ib mice reduced SIRT1 expression. LKB1 overexpression reduced hepatic triglyceride levels, linking LKB1/AMPK signaling to SIRT1 expression. Taken together, our data show that G6PT deficiency leads to impaired autophagy in GSD-Ib primarily by downregulating SIRT1/FoxO3a/LKB1/AMPK signaling [Reference 1].
Additional Funding
- The Children’s Fund for Glycogen Storage Disease Research
- CRISPR Therapeutics (Cambridge, MA), under a cooperative research and development agreement (CRADA)
- Beam Therapeutics, Inc (Cambridge, MA), under a CRADA
Publications
- Arnaoutova I, Zhang L, Chen HD, Mansfield BC, Chou JY. Correction of metabolic abnormalities in a mouse model of glycogen storage disease type Ia by CRISPR/Cas9-based gene editing. Mol Ther 2021;29(4):1602–1610.
- Zhang L, Cho JH, Arnaoutova I, Mansfield BC, Chou JY. An evolutionary approach to optimizing glucose-6-phosphatase-alpha enzymatic activity for gene therapy of glycogen storage disease type Ia. J Inherit Metab Dis 2019;42:470–479.
- Zhang L, Lee C, Arnaoutova I, Anduaga J, Starost MF, Mansfield BC, Chou JY. Gene therapy using a novel G6PC-S298C variant enhances the long-term efficacy for treating glycogen storage disease type Ia. Biochem Biophys Res Commun 2020;527:824–830.
- Gautam S, Zhang L, Arnaoutova I, Mansfield BC, Chou, JY. The signaling pathways implicated in impairment of hepatic autophagy in glycogen storage disease type Ia. Hum Mol Genet 2020;29:834–844.
Collaborators
- Alessandra Eva, PhD, Istituto Giannina Gaslini, Genova, Italy
- Brian C. Mansfield, PhD, Foundation Fighting Blindness, Columbia, MD
- Matthew F. Starost, PhD, Diagnostic & Research Services Branch, Division of Veterinary Resources, NIH, Bethesda, MD
- David A. Weinstein, MD, MSc, University of Connecticut School of Medicine, Farmington, CT
Contact
For more information, email chou@helix.nih.gov or visit https://irp.nih.gov/pi/janice-chou.