Molecular Genetics of an Imprinted Gene Cluster on Mouse Distal Chromosome 7

- Karl Pfeifer, PhD, Head, Section on Epigenetics
- Claudia Gebert, PhD, Biologist
- Beenish Rahat, PhD, Postdoctoral Fellow
- Hyungchul Lee, PhD, Visiting Fellow
- Colter Brainard, BS, Postbaccalaureate Fellow
- Rachel Muti, BS, Postbaccalaureate Fellow
Genomic imprinting is an unusual form of gene regulation by which an allele’s parental origin restricts allele expression. For example, almost all expression of the noncoding RNA tumor-suppressor gene H19 is from the maternal chromosome. In contrast, expression of the neighboring Insulin-like Growth Factor 2 gene (Igf2) is from the paternal chromosome. Imprinted genes are not randomly scattered throughout the chromosome but rather are localized in discrete clusters, where monoallelic expression is regulated by a common cis-acting DNA–regulatory element called the Imprinting Control Region (ICR). We study a cluster of imprinted genes on the distal end of mouse chromosome 7 (Figure 1). The syntenic region in humans (11p15.5) is highly conserved in gene organization and expression patterns. Imprinting of H19 and of Igf2 is regulated by the H19 ICR, which is located just upstream of the H19 promoter. We showed that the molecular function of the H19 ICR is to organize the region into alternative 3D structures. In humans, epigenetic mutations that disrupt H19 ICR function result in loss of monoallelic expression. Mutations in the paternal H19 ICR lead to loss of Igf2 expression and biallelic (2X) H19 expression and are associated with the Russell-Silver syndrome. Mutations in the maternal H19 ICR lead to loss of H19 but biallelic (2x) Igf2 expression and are associated with the Beckwith-Wiedemann syndrome and several pediatric cancers. Our lab generated mouse models that phenocopy the human diseases, and our goal is to characterize the molecular defects associated with mis-expression of Igf2/H19 and to understand how these molecular defects lead to disease and cancer. In particular, we strive to understand the role of development in disease progression.
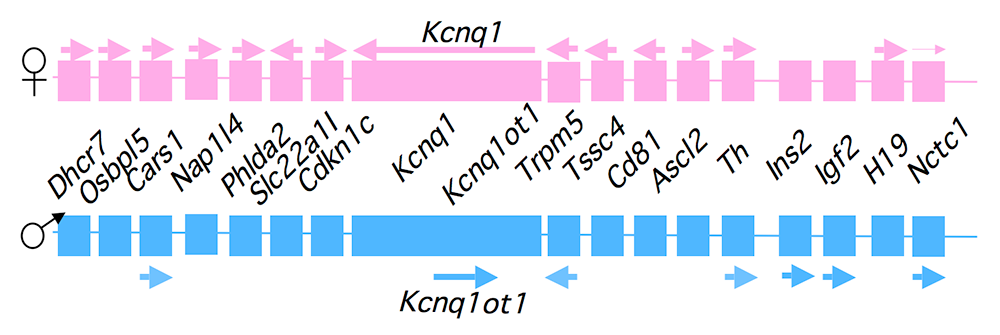
Figure 1. An imprinted domain on mouse distal chromosome 7
Click image to view.
Maternal (pink) and paternal (blue) chromosomes are indicated. Horizontal arrows denote RNA transcription.
Alternative long-range interactions between distal regulatory elements establish allele-specific expression at the Igf2/H19 locus.
Paternally expressed Igf2 lies about 80 kb upstream of the maternal-specific H19 gene. Using cell-culture systems as well as transgene and knockout experiments in vivo, we identified the enhancer elements responsible for activation of the two genes. The elements are shared and are all located downstream of the H19 gene (Figure 2). As mentioned above, imprinting at the Igf2/H19 locus depends on the 2.4 kb H19 ICR, which lies between the two genes, just upstream of the H19 promoter (Figure 2). On the maternal chromosome, binding of the CTCF protein, a transcriptional repressor, to the H19 ICR establishes a transcriptional insulator that organizes the chromosome into loop structures that bring the H19 promoter into contact with downstream enhancers but exclude the Igf2 promoter from these enhancer interactions. The loops favor H19 expression but block interactions between the maternal Igf2 promoters and the downstream shared enhancers, thus preventing maternal Igf2 expression. Upon paternal inheritance, the cytosine residues within the ICR DNA sequences are methylated, which prevents binding of the CTCF protein, so that a transcriptional insulator is not established. Thus, paternal Igf2 promoters and the shared enhancers interact via DNA loops, and expression of paternal Igf2 is facilitated. Taken together, we find that the fundamental role of the ICR is to organize the chromosomes into alternative 3-D configurations that promote or prevent expression of the Igf2 and H19 genes.
The H19 ICR is not only necessary but is also sufficient for genomic imprinting. To demonstrate this, we used knock-in experiments to insert the 2.4 kb element at heterologous loci and demonstrated its ability to imprint these regions. Furthermore, analyses of the loci confirmed and extended the transcriptional model described above. Upon maternal inheritance, even ectopic ICR elements remain unmethylated, bind to the CTCF protein, and form transcriptional insulators. Paternally inherited ectopic ICRs become methylated, cannot bind to the CTCF, and therefore promote alternative loop domains distinct from those organized on maternal chromosomes. Most curious was the finding that DNA methylation of ectopic ICRs is not acquired until relatively late in development, after the embryo implants in the uterus. In contrast, at the endogenous locus, ICR methylation occurs during spermatogenesis. The findings thus imply that DNA methylation is not the primary imprinting mark that distinguishes maternally from paternally inherited ICRs.
The Nctc1 gene lies downstream of H19 and encodes a spliced, polyadenylated long noncoding RNA (lncRNA), which is transcribed across the muscle enhancer element (ME in Figure 2), which is shared by Igf2 and H19. Nctc1 expression depends on this enhancer element. Concordantly, the shared enhancer interacts with the Nctc1 promoter, just as it interacts with the maternal H19 and paternal Igf2 promoters. We showed that all three co-regulated promoters (Igf2, H19, and Nctc1) also physically interact with each other in a manner that depends on their interactions with the shared enhancer. Thus, enhancer interactions with one promoter do not preclude interactions with another promoter. Moreover, we demonstrated that such promoter-promoter interactions are regulatory; they explain the developmentally regulated imprinting of Nctc1 transcription. Taken together, our results demonstrate the importance of long-range enhancer-promoter and promoter-promoter interactions in physically organizing the genome and establishing the gene expression patterns that are crucial for normal mammalian development.
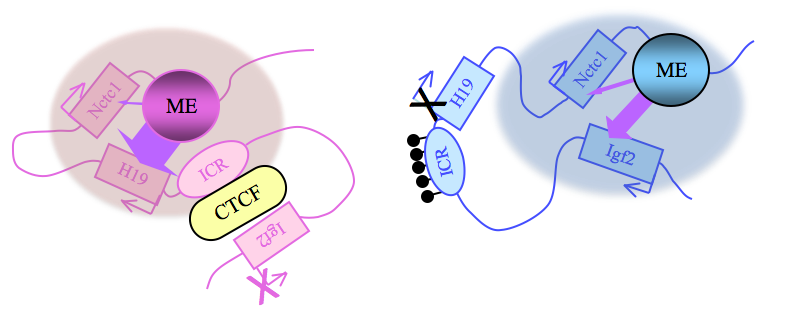
Figure 2. Distinct maternal and paternal chromosomal conformations at the distal 7 locus
Click image to view.
Epigenetic modifications on the 2.4 kb ICR generate alternative 3D organizations across a large domain on paternal (blue) and maternal (pink) chromosomes and thereby regulate gene expression. ICR, imprinting control region; ME, muscle enhancer; filled lollipops, CpG methylation covering the paternal ICR.
Molecular mechanisms for tissue-specific promoter activation by distal enhancers
Normal mammalian development is absolutely dependent on establishing the appropriate patterns of expression of thousands of developmentally regulated genes. Most often, development-specific expression depends on promoter activation by distal enhancer elements. The Igf2/H19 locus is a highly useful model system for investigating mechanisms of enhancer activation. First, the biological significance of the model is clear, given that expression of these genes is so strictly regulated. Even twofold changes in RNA levels are associated with cancer and developmental disorders. Second, we already know much about the enhancers in this region and have established powerful genetic tools to investigate their function. Igf2 and H19 are co-expressed throughout embryonic development and depend on a series of tissue-specific enhancers that lie between 8 and more than 150 kb downstream of the H19 promoter (or between 88 and more than 130 kb downstream of the Igf2 promoters). The endodermal and muscle enhancers have been precisely defined, and we generated mouse strains carrying deletions that completely abrogate enhancer function. We also generated insulator insertion mutations that specifically block muscle enhancer activity. We used these strains to generate primary myoblast cell lines so that we can combine genetic, molecular, biochemical, and genomic analyses to understand the molecular bases for enhancer functions.
The lncRNA, encoded by Nctc1, is an essential element of the muscle enhancer, as demonstrated by transient transfection analyses that define a 300–bp element that is both necessary and sufficient for maximal enhancer activity. However, stable transfection and mouse mutations indicate that this core element is not sufficient for enhancer function in a chromosomal context. Instead, the Nctc1 promoter element is also essential; the Nctc1 RNA itself is not required (at least in trans). Instead mutational analysis demonstrates that it is Nctc1 transcription through the core enhancer that is necessary for enhancer function. Curiously, the Nctc1 promoter has chromatin features typical of both a classic enhancer and a classic peptide-encoding promoter. Several recent genomic studies also suggested a role for noncoding RNAs in gene regulation and enhancer function. We will use our model system to characterize the role of Nctc1 transcription in establishing enhancer orientation, enhancer promoter specificity, and enhancer tissue specificity.
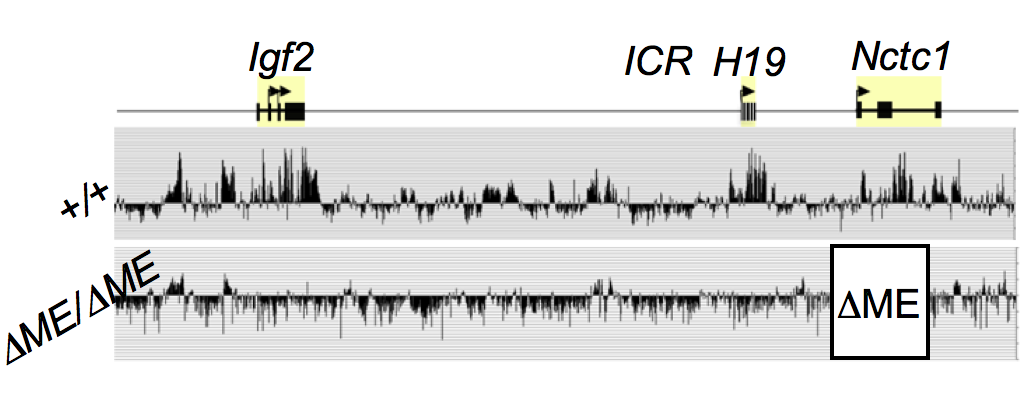
The muscle enhancer (ME) directs RNA polymerase (RNAP) II not only to its cognate promoters (i.e., to the H19 and Igf2 promoters) but also across the entire intergenic region. To demonstrate this, we used ChIP-on-chip to analyze RNAP localization on chromatin prepared from wild-type and enhancer-deletion (DME) cell lines (Figure 3). As expected, RNAP binding to the H19 and Igf2 promoters is entirely enhancer-dependent. Curiously, we also noted enhancer-dependent RNAP localization across the entire locus, including the large intergenic domain between the two genes. Furthermore, RNAP binding is associated with RNA transcription. Thus, the enhancer regulates accessibility and RNAP binding not only at specific localized sites but across the entire domain. The results support a facilitated tracking model for enhancer activity.
Figure 3.
Click image to view.
The shared muscle enhancer (ME) directs RNAP binding and RNA transcription across the entire 150 kb locus.
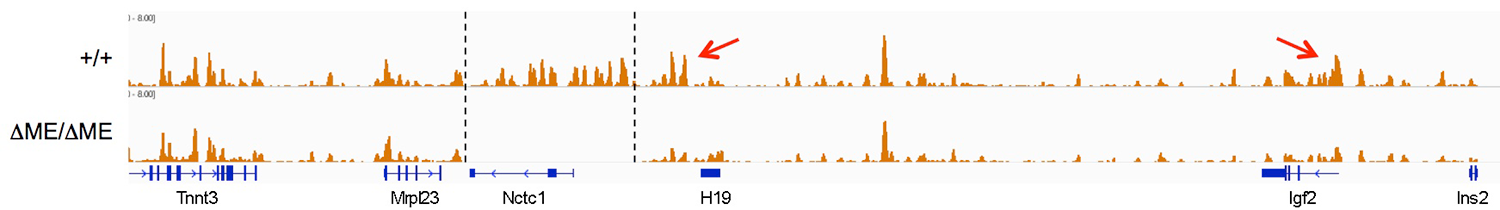
RNAP binding at ‘real’ genes and across the intergenic regions is qualitatively different. To demonstrate this, we used naturally occurring single-nucleotide polymorphisms (SNPs) to investigate allelic differences in binding of RNAP and activation of gene expression in wild-type cells and in cells carrying enhancer deletions or insulator insertion mutations. RNAP binding across the Igf2 and H19 genes is both enhancer-dependent and insulator-sensitive; that is, a functional insulator located between an enhancer and its regulated gene prevents RNAP binding and likewise prevents RNA transcription. Across the intergenic regions, RNAP binding and RNA transcription are similarly enhancer-dependent (see above). However, intergenic RNAP binding and transcription are not insulator-sensitive. The results indicate that insulators do not serve solely as a physical block for RNAP progression, but rather they specifically interfere with certain RNAP states or activities.
The muscle enhancer regulates RNAP binding and RNA transcription, but does not establish chromatin structures, because both RNA transcription and RNAP binding across the Igf2/H19 domain are entirely dependent upon the muscle enhancer. For example, levels of H19 RNA are reduced more than 10,000-fold in muscle cells in which the enhancer has been deleted. To test the dependence of chromatin structure on enhancer activity, we performed ChIP-Seq on wild-type and on enhancer-deletion cell lines using antibodies against the histones H3K4me1, H3K43me3, and H3K36me3. Surprisingly, we saw no changes in the patterns of chromatin modification (Figure 4). Thus, a functional enhancer and active RNA transcription are not important for establishing chromatin structures at the Igf2/H19 domain.
Click image to view.
Figure 4. Chromatin patterns at the Igf2/H19 locus are independent of enhancer activity.
Chromatin was isolated from wild-type and enhancer-deletion muscle cells, using antibodies against H3K4me1, and analyzed by DNA sequencing.
Functions of H19 lncRNA in regulating cell-cycle progression and senescence
To determine the biochemical functions of H19 lncRNA, we use in vitro models including primary myoblasts, C2C12 myoblasts, and NIH3T3 cells. Abrupt depletion of H19 by either siRNA or cre-induced recombination of H19–floxed alleles results in increased p21 RNA (p21 is a cyclin-dependent kinase inhibitor involved in cell-cycle arrest) and peptide, and such increased p21 activity in turn prevents cell-cycle progression and induces cellular senescence. H19 lncRNA regulation of p21 is at the level p21 mRNA stability and translation efficiency and occurs via the p21 3′-UTR. Genetic and biochemical analyses suggest that H19 lncRNA facilitates interactions between p21 mRNA and Wig1. Current experiments focus on identifying the molecular mechanisms for these regulatory actions.
Functions of H19 lncRNA in regulating cardiac development
The Beckwith-Wiedemann syndrome (BWS) is a developmental disorder characterized by generalized overgrowth of the fetus and a high risk for several neonatal cancers. Many BWS patients also display cardiac problems. BWS can be explained by one of two different genetic lesions: loss of function of the CDKN1C gene or maternal loss of imprinting at the H19/Igf2 locus. Maternal loss of imprinting has the effect of doubling Igf2 expression while concomitantly reducing H19 RNA levels. Curiously, children born via artificial reproductive technology (ART) show increased incidence of BWS, which can be explained by increasingly frequent loss of H19/Igf2 imprinting in such children. Moreover the children show high frequency of cardiac dysfunction. Taken together, such findings suggest that abnormal expression of the H19/Igf2 locus can lead to cardiac problems.
Figure 5. Cardiac disease in H19–deficient mice
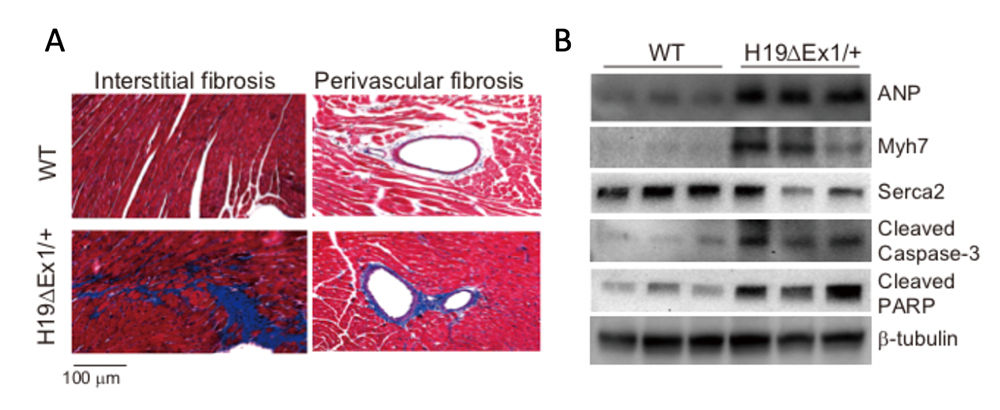
Click image to view.
Mice lacking H19 are hypertrophic, fibrotic (panel A), display protein expression profiles typical of cardiac failure (panel B), and show aberrant function on echocardiograms.
We observed that our BWS mouse model also results in cardiac dysfunction, as measured by echocardiography and ECG analyses. Molecular and molecular-genetic analyses demonstrate that biallelic Igf2 and loss of H19 play independent and distinct roles in generating the BWS phenotype. Biallelic expression of Igf2 results in elevated levels of circulating IGF2 peptide, which super-activates insulin and insulin-like receptor kinases in cardiomyocytes, resulting in hyper-activation of AKT/mTOR signaling pathways, which in turn causes cardiomyocyte hypertrophy and hyperplasia. Such effects result in a cardiac hypertrophy that is non-pathologic and transient, i.e., the hearts function normally and, as long as H19 levels are normal, the heart size normalizes after birth once Igf2 expression is repressed. Thus, there are no significant health effects associated with loss of imprinting of Igf2 only.
Loss of expression of H19 is pathologic (Figure 4). Hearts show progressive heart disease, as manifested by hypertrophy, increased fibrosis, expression of cardiac failure markers, and reduced and abnormal heart function, as measured by echocardiography. H19 expression in hearts is restricted to endothelial cells. In vivo analyses of whole hearts and in vitro analyses of isolated endothelial cells show that reduction in H19 results in increased endothelial-to-mesenchymal transition (EMT). EMT is an essential feature of normal cardiac development; for example, formation of cardiac valves requires EMT. However, elevated frequency of EMT is associated with heart disease. Our data support the notion that H19 regulates the cell fate of endothelial cells, and future experiments aim to identify the underlying molecular mechanisms.
The H19 gene does not encode a protein. Rather H19 RNA is the functional gene product. The H19 precursor RNA is processed into two final products: a 2.3 kb lncRNA and microRNA 675. To determine which H19 RNA is essential to prevent cardiomyopathy, we developed two new mouse strains that selectively disrupt accumulation of the lncRNA or the miRNA. Disruption of the lncRNA is necessary and sufficient to induce cardiac disease. To understand the molecular bases for H19 lncRNA function, we generated alleles that disrupt specific domains. We observe that disruption of let7 miRNA binding sites on the H19 RNA results in the same myopathies as complete gene ablation (Figure 6).
Figure 6. Let7 binding sites on H19 lncRNA are essential for normal heart function.
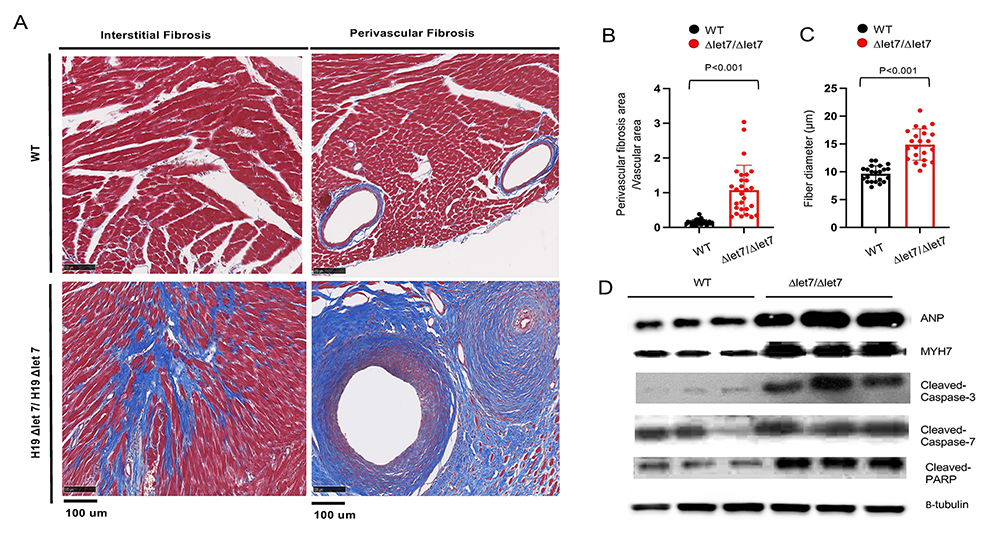
Click image to view.
Mice carrying H19 alleles that do not include let7 binding sites are fibrotic (panels A and B), hypertrophic (panel B), and display protein expression profiles typical of cardiac failure.
Publications
- Park K-S, Rahat B, Lee HC, Noeker J, Mitra A, Kean CM, Knutsen RH, Springer D, Gebert CM, Kozel BA, Pfeifer K. Cardiac pathologies in mouse loss of imprinting models are due to misexpression of H19 long noncoding RNA. eLife 2021;10:e67250.
- Flores DJ, Duong T, Brandenberger LO, Mitra A, Shirali A, Johnson JC, Springer D, Noguchi A, Yu ZX, Ebert SN, Ludwig A, Knollmann BC, Levin MD, Pfeifer, K. Conditional ablation and conditional rescue models for Casq2 elucidate the role of development and of cell-type specific expression of Casq2 in the CPVT2 phenotype. Hum Mol Genet 2018;27:1533–1544.
- Park K-S, Mitra A, Rahat B, Kim B, Pfeifer K. Loss of imprinting mutations define both distinct and overlapping roles for misexpression of IGF2 and of H19 lncRNA. Nucleic Acids Res 2017;45:12766–12779.
- Mitra A, Park K-S, Pfeifer K. Circular RNAs and competing endogenous (ceRNA) networks. Transl Cancer Res 2018;7:S624–628.
- Geng H, Bu HF, Liu F, Wu L, Pfeifer K, Chou PM, Wang X, Sun J, Lu L, Pandey A, Bartolomei MS, De Plaen IG, Wang P, Yu J, Qian J, Tan XD. In inflamed intestinal tissues and epithelial cells, interleukin 22 signaling increases expression of H19 long noncoding RNA, which promotes mucosal regeneration. Gastroenterology 2018;155:144–155.
Collaborators
- Judy Kassis, PhD, Section on Gene Expression, NICHD, Bethesda, MD
- Beth Kozel, MD, PhD, Cardiovascular & Pulmonary Branch, NHLBI, Bethesda, MD
- Danielle A. Springer, VMD, Dipl ACLAM, Animal Program, NHLBI, Bethesda, MD
Contact
For more information, email kpfeifer@helix.nih.gov or visit https://pfeiferlab.nichd.nih.gov.