Virulence Mechanisms of Microbial Pathogens

- Matthias Machner, PhD, Head, Section on Microbial Pathogenesis
- Devanand Bondage, PhD, Visiting Fellow
- Katherine Bonnington, PhD, Visiting Fellow
- Candice Chang, PhD, Visiting Fellow
- Nicole Ellis, PhD, Visiting Fellow
- Stephanie Lehman, PhD, Visiting Fellow
- Xiao Li, PhD, Visiting Fellow
- Sabrina Bouchard, BS, Postbaccalaureate Student
- Jacob Olondo-Kuba, BS, Postbaccalaureate Student
Our main research goal is to define mechanisms by which pathogenic bacteria subvert the human host defense and cause disease. In parallel, we investigate whether these mechanisms may be manipulated for preventative and/or therapeutic purposes. As a model organism we use the bacterium Legionella pneumophila, the causative agent of a potentially fatal respiratory infection known as Legionnaires' disease. According to the CDC (Centers for Disease Control), the number of Legionnaires' disease cases in the U.S. has risen more than four-fold over the past 15 years, making L. pneumophila an emerging pathogen of increasing relevance. Contrary to what its name may imply, Legionnaires’ disease occurs in individuals of all ages, including children who receive respiratory therapy, newborns who had recently undergone surgery or under-water birth, and children who are immune-compromised. We are committed to an in-depth analysis of the mechanisms that allow L. pneumophila to exploit the human host and cause disease. Insights gained from our studies will ultimately improve our ability to diagnose, prevent, and fight Legionnaires’ disease and related illnesses, thereby contributing to the success of NICHD’s mission.
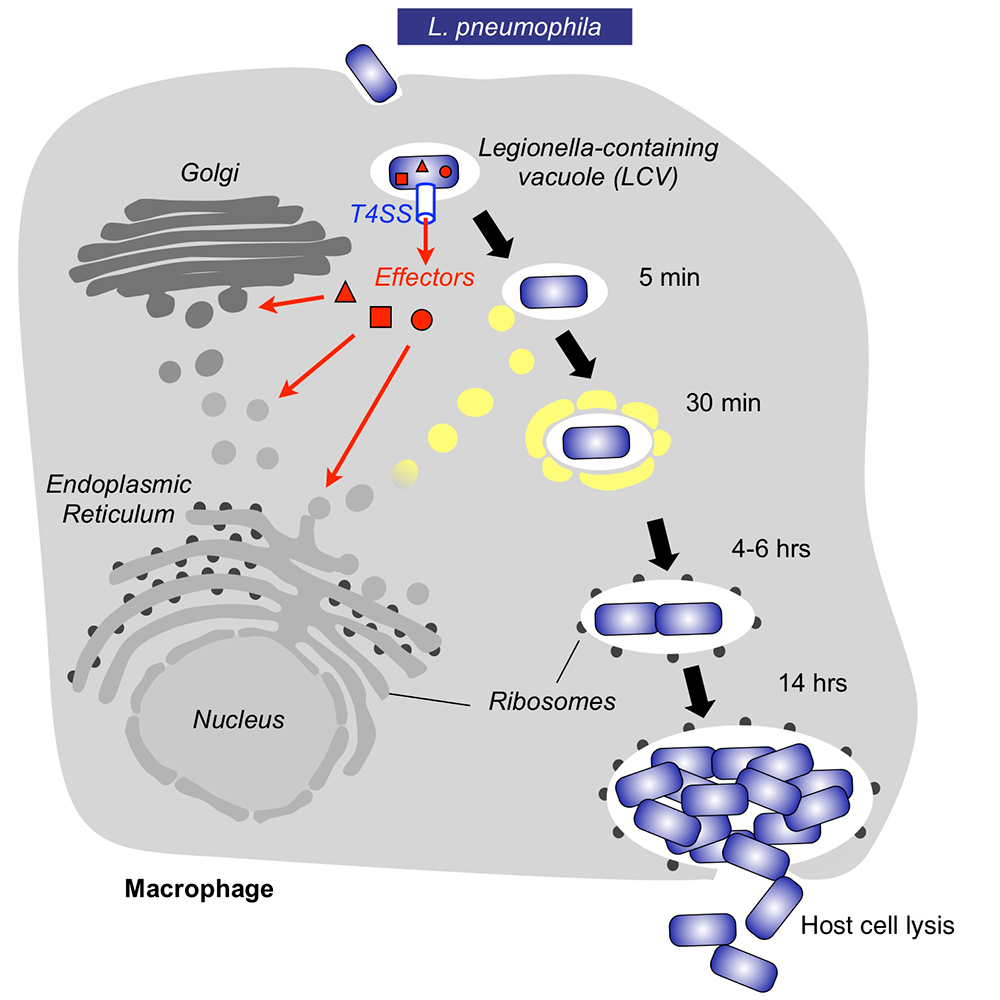
Within freshwater environments, L. pneumophila exists as an intracellular parasite of single-cell organisms know as amoeba. Upon inhalation of contaminated water droplets, L. pneumophila enters the lung and is phagocytosed (taken up) by specialized immune cells known as alveolar macrophages (Figure 1). Instead of being degraded by these cells, the pathogen establishes a protective membrane compartment: the Legionella-containing vacuole (LCV). Within this intravacuolar niche, L. pneumophila can replicate to high numbers before killing the host cell and infecting neighboring cells.
The virulence of L. pneumophila relies on the activity of close to 300 proteins, or effectors, that are delivered into the host cytosol by a specialized translocation apparatus called the Dot/Icm type IV secretion system (T4SS) (Figure 1). L. pneumophila mutants with a non-functional T4SS are degraded by macrophages, underscoring the importance of the translocated effectors for host-cell manipulation and bacterial virulence.
Our main objective is to obtain detailed mechanistic insight into L. pneumophila effectors by investigating their biological role at molecular, cellular, and structural levels. Knowledge obtained from these studies can help in the development of novel therapeutics aimed at treating or preventing Legionnaires' disease and related illnesses.
Figure 1. Intracellular replication cycle of Legionella pneumophila
Click image to view.
Upon uptake by a macrophages, L. pneumophila delivers a large number of effector proteins (red) through the Dot/Icm type IV secretion system (T4SS) into the host cytosol. The effectors manipulate signaling and trafficking pathways in order to establish conditions favorable for L. pneumophila growth. Eventually, the host cell is lyzed, and L. pneumophila bacteria infect neighboring cells.
The Legionella effector kinase LegK7 hijacks the host Hippo pathway to promote infection.
Microbial pathogens manipulate host-cell signaling pathways by encoding molecular mimics of protein kinases. Kinases catalyze target phosphorylation by attaching a phosphate group onto side chains of substrate proteins, preferentially serine, threonine, or tyrosine residues, thereby altering the activity, localization, or stability of their substrates. Protein phosphorylation is one of the most abundant and most important post-translational modifications in living cells, and mammals encode hundreds of kinases that target thousands of substrate proteins. To promote infection, many pathogens, including bacteria and eukaryotic parasites, encode effector kinases that are translocated into the host cytosol, where they alter signaling cascades, suggesting that kinase mimicry is a commonly used strategy to exploit host cell function.
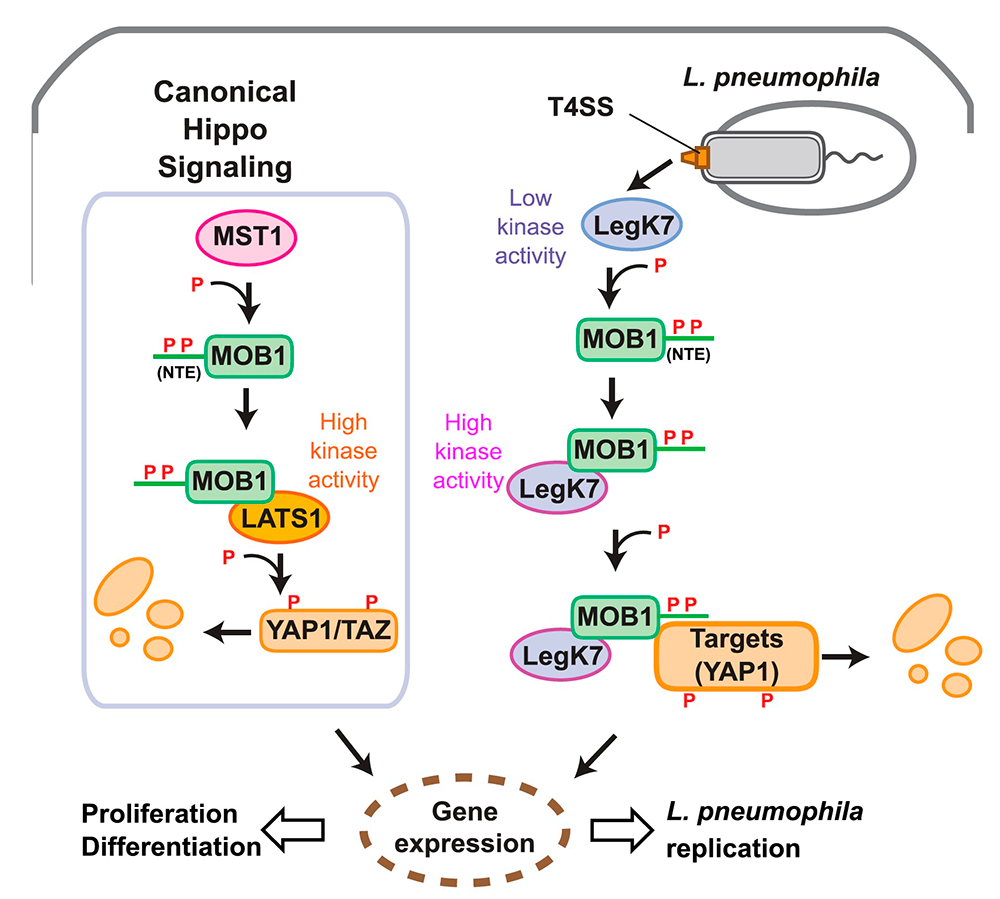
Using structural prediction to identify cryptic catalytic domains within L. pneumophila effectors, we discovered that the protein LegK7 contains a central domain (residues 183–462) that has folding homology to eukaryotic protein kinases. Using a specific labeling technique, we experimentally confirmed that LegK7 indeed exhibits kinase activity in vitro. Upon probing a human protein microarray platform with LegK7, we identified MOB1 as a direct substrate of LegK7 (Figure 2). MOB1 is a key scaffold protein within the Hippo kinase signaling pathway, which, in eukaryotes, controls cell-cycle progression, cell proliferation and differentiation, and apoptosis. In a variety of in vitro and cell-based assays, we subsequently showed that LegK7 hijacks the Hippo pathway by molecularly mimicking the host Hippo kinase (MST1 in mammals), the key regulator of pathway activation. LegK7, like Hippo/MST1, phosphorylates the scaffolding protein MOB1 on two residues, which triggers a signaling cascade, resulting in the degradation of the transcriptional regulators TAZ and YAP1. Transcriptome analysis by RNA-Seq revealed that LegK7–mediated targeting of TAZ and YAP1 alters the transcriptional profile of mammalian macrophages, a key cellular target of L. pneumophila infection. Specifically, genes targeted by the transcription factor PPARg, which is regulated by TAZ, displayed altered expression, and continuous interference with PPARg activity rendered macrophages less permissive to L. pneumophila intracellular growth. Thus, a conserved L. pneumophila effector kinase exploits the Hippo pathway to promote bacterial growth and infection (Figure 2) [Reference 1].
Figure 2. Legionella LegK7 manipulates the host cell Hippo signaling pathway.
Click image to view.
Cartoon comparing canonical Hippo signaling (left) with signaling by Legionella LegK7 (right). In the canonical Hippo pathway, the Hippo kinase MST1 phosphorylates the N-terminal extension (NTE) of MOB1A on two tyrosine residues, T12 and T35, to promote formation of the MOB1A–LATS1 kinase complex. The activated MOB1A–LATS1 kinase complex phosphorylates the transcriptional regulators YAP1/TAZ, which results in degradation of YAP1/TAZ and, consequently, expression of genes involved in cell proliferation or differentiation.
During infection, L. pneumophila translocates the effector kinase LegK7 into host cells. Like MST1, LegK7 directly phosphorylates T12 and T35 on the NTE of MOB1A. LegK7 then interacts directly with phosphorylated MOB1A and becomes highly active. Active LegK7 uses MOB1A to recruit downstream substrates, possibly YAP1, for further phosphorylation and manipulates the conserved Hippo pathway to promote infection.
Legionella LegK7 exploits the host scaffold protein MOB1A for allostery and substrate phosphorylation.
The finding that LegK7 targets MOB1A and manipulates its function points toward an unexpected role for the Hippo pathway in microbial pathogenesis. Given that LegK7 appears to functionally mimic MST1/2, it became important to more thoroughly investigate this host-pathogen interaction and to determine, at a molecular level, how LegK7 exploits a signaling pathway that is engrained within the developmental program of eukaryotic cells.
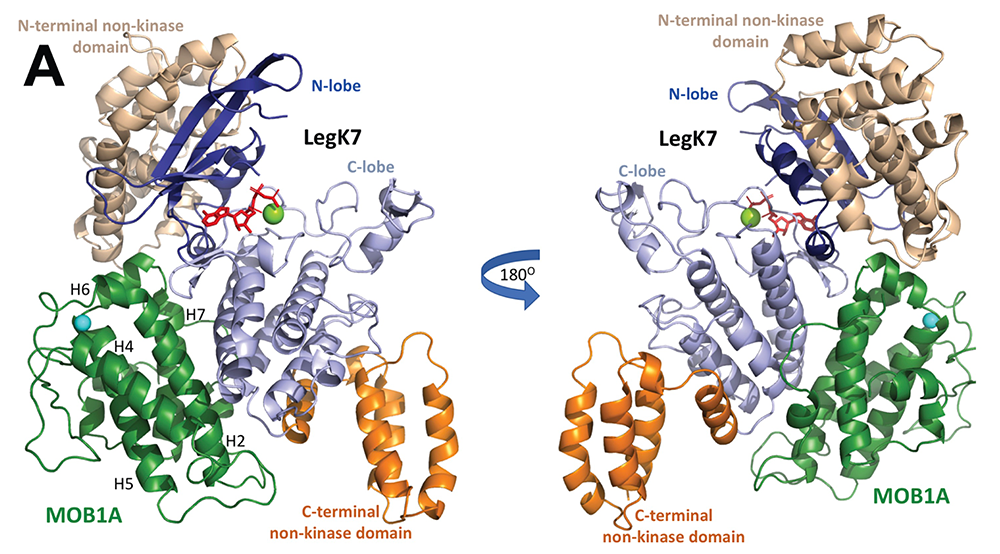
Using in vitro kinase reconstitution assays, we showed that MOB1A, in addition to being a substrate of LegK7, functions as an allosteric activator of LegK7's kinase activity. A crystallographic analysis of the LegK7–MOB1A complex revealed that the N-terminal half of LegK7 is structurally similar to eukaryotic protein kinases, and that MOB1A, like the mammalian kinase LATS1/2, directly binds to the LegK7 kinase domain (Figure 3). Substitution of interface residues critical for complex formation abrogated allosteric activation of LegK7 both in vitro and within cells and diminished MOB1A phosphorylation. We also provided evidence that LegK7 forms a complex with MOB1A in order to use its N-terminal extension as a binding platform for the recruitment of downstream substrates, so that their phosphorylation can occur more efficiently.
Together, our findings suggest that LegK7 has evolved into a functional chimera of MST1/2 and LATS, and that it combines features from both of the core kinases of the Hippo pathway in order to exploit the downstream signaling cascade. We propose that, by using this strategy, L. pneumophila has developed the ability to survive within a wide range of diverse host species, including humans [Reference 2].
Figure 3. Crystal structure of the LegK7–MOB1A complex
Click image to view.
Cartoon representation of the LegK7(11–530)–MOB1A33 complex. MOB1A33 (green) contacts predominantly the kinase domain of LegK7 with additional contacts to the N-terminal domain. Flanking N- (wheat) and C-terminal domains of LegK7 (orange); N lobe of the kinase domain (blue); C lobe (slate); adenosine monophosphate (AMP)-PNP (red, in stick representation); Mn2+ (green sphere).
A multiplex CRISPR interference tool for virulence gene interrogation in an intracellular pathogen
Clustered, regularly interspaced short palindromic repeats (CRISPR)-Cas gene editing technologies have recently arisen as a mechanism for both fast and targeted gene manipulation in a variety of systems. In the simplest CRISPR-Cas system, Type II, only a single protein known as Cas9 is required for crRNA–guided DNA cleavage, making it the most developed genetic tool. In the absence of target cleavage, catalytically inactive dCas9 imposes transcriptional gene repression by sterically precluding RNA polymerase activity at a given gene to which it was directed by CRISPR (cr)RNAs. This gene-silencing technology, referred to as CRISPR interference (CRISPRi), has been employed in various bacterial species to interrogate genes, mostly individually or in pairs. To our knowledge, no group has fully exploited or probed the natural multiplex capability of CRISPR repeat/spacer arrays as a gene silencing tool in bacterial systems.
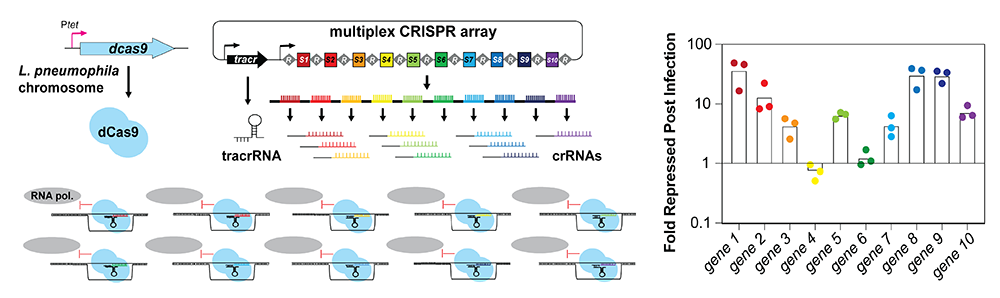
We thus developed a multiplex (MC) CRISPRi platform in the pathogen L. pneumophila capable of silencing up to ten genes simultaneously (Figure 4). We provided proof-of-concept that this novel platform can be used not only during growth in axenic media but also during macrophage infection, where it reproduced known intracellular growth phenotypes. Importantly, by placing the crRNA–encoding spacer in positions further downstream within the array, the degree of gene silencing was titratable. In contrast, when combined with a boxA element (a short DNA motif that antagonizes premature transcript termination in bacteria by recruiting the anti-termination factor Nus), our 10-plex CRISPR array had the potential to silence up to ten unique genes simultaneously (Figure 4), making it a powerful tool to study even synergistic genetic interactions [Reference 3].
Looking forward, the multiplex CRISPRi approach we developed holds the promise of one day probing functional overlap amongst the hundreds of L. pneumophila effectors. Not only can genes be silenced in bulk groups during infection, but the mobility of our single plasmid–based CRISPRi platform allows for easy transfer of MC constructs into a variety of Legionella mutant-strain backgrounds to directly assess redundancy, presuming they have been equipped with a copy of dCas9.
Figure 4. Concept of multiplex CRISPR interference (CRISPRi)
Click image to view.
A series of repeats, R, and spacers, S1-S10, are expressed as a single precursor crRNA. Upon processing, individual crRNAs come together with a tracrRNA and dCas9 to simultaneously target ten unique genes for silencing. Fold repression of target genes 1–10 compared with a control strain was determined using qPCR.
Structural insight into the membrane-targeting domain of the Legionella deAMPylase SidD
AMPylation, the post-translational modification with adenosine monophosphate (AMP), is catalyzed by effector proteins from a variety of pathogens. L. pneumophila is thus far the only known pathogen that, in addition to encoding an AMPylase (SidM), also encodes a deAMPylase, called SidD, that reverses SidM–mediated AMPylation of the host-cell vesicle transport GTPase Rab1; deAMPylation is catalyzed by the N-terminal phosphatase-like domain of SidD. The structure of the C-terminal domain and its biological role had remained unclear.
We determined the crystal structure of full length SidD, including the previously uncharacterized C-terminal domain (Figure 5). We found that the C-terminal domain assumed a novel fold not yet described in other proteins, which is critical for membrane localization of SidD within host cells. Notably, the localization depended on two structural elements within the C-terminal domain: a flexible loop composed of hydrophobic amino acid residues, which represents a general membrane-targeting determinant; and a two-helix bundle at the C-terminal end that functions as the specificity determinant. Deletion of either of these elements interfered with the correct localization of SidD within mammalian cells. Moreover, we found that, during growth in mouse macrophages, a L. pneumophila strain producing SidD without the loop phenocopied a L. pneumophila strain producing no SidD at all, thus revealing that membrane targeting of SidD via its C-terminal domain is a critical prerequisite for its ability to catalyze Rab1 deAMPylation during L. pneumophila infection. Future studies will reveal whether additional effectors from L. pneumophila or related pathogens use a similar membrane-targeting strategy within host cells [Reference 4].
Figure 5. SidD membrane localization requires the C-terminal domain.

Click image to view.
A. Domain organization of SidD with the N-terminal domain (NTD, blue) and C-terminal domain (CTD, magenta). The hydrophobic loop (dashed line) is unstructured, indicative of intrinsic flexibility. Numbers indicate amino acid positions.
B. Intracellular localization of CTD variants. Transiently transfected COS-1 cells producing either GFP (control) or GFP-CTD variants were fixed and stained using an antibody directed against the Golgi marker protein giantin (middle). Merged images of SidD proteins (green) and giantin (red). Scale bar: 10 μm.
C. CTD(322–450) localizes to mitochondria membranes. Transiently transfected COS-1 cells coproducing GFP-CTD(322–450) and Mito-RFP (a mitochondria marker) were chemically fixed, and the fluorescence signal was examined by confocal microscopy. Arrowheads indicate the position of membranes magnified in the insets. Scale bar: 10 μm.
Publications
- Lee PC, Machner MP. The Legionella effector kinase LegK7 hijacks the host Hippo pathway to promote infection. Cell Host Microbe 2018;24:429–438.
- Lee PC, Beyrakhova K, Xu C, Boniecki MT, Lee MH, Onu CJ, Grishin AM, Machner MP, Cygler M. The Legionella kinase LegK7 exploits the Hippo pathway scaffold protein MOB1A for allostery and substrate phosphorylation. Proc Natl Acad Sci USA 2020;117:14433–14443.
- Ellis NA, Kim B, Tung J, Machner MP. A multiplex CRISPR interference tool for virulence gene interrogation in Legionella pneumophila. Commun Biol 2021;4:157.
- Tascón I, Li X, Lucas M, Nelson D, Vidaurrazaga A, Lin YH, Rojas AL, Hierro A, Machner MP. Structural insight into the membrane targeting domain of the Legionella deAMPylase SidD. PLoS Pathog 2020;16(8):e1008734.
Collaborators
- Miroslaw Cygler, PhD, University of Saskatchewan College of Medicine, Saskatoon, Canada
- Robert Heinzen, PhD, Coxiella Pathogenesis Section, Rocky Mountain Laboratories, NIAID, Hamilton, MT
- Aitor Hierro, PhD, CIC bioGUNE Institute, Bilbao, Spain
- Michal Jarnik, PhD, Section on Intracellular Protein Trafficking, NICHD, Bethesda, MD
- Anton Simeonov, PhD, Division of Pre-Clinical Innovation, NCATS, Bethesda, MD
- Chad Williamson, PhD, Advanced Microscope Facility, NICHD, Bethesda, MD
Contact
For more information, email machnerm@mail.nih.gov or visit https://machnerlab.nichd.nih.gov.