Protein Sorting in the Endomembrane System

- Juan S. Bonifacino, PhD, Head, Section on Intracellular Protein Trafficking
- Rafael Mattera, PhD, Staff Scientist
- Xiaolin Zhu, Nurse, Technician
- Carlos M. Guardia, PhD, Visiting Fellow
- Rui Jia, PhD, Visiting Fellow
- Raffaella De Pace, PhD, Visiting Fellow
- Saikat Ghosh, PhD, Visiting Fellow
- Tal Keren-Kaplan, PhD, Visiting Fellow
- Morie Ishida, PhD, Visiting Fellow
- Elodie Mailler, PhD, Visiting Fellow
- Amra Saric, PhD, Visiting Fellow
- Ganesh Shelke, PhD, Collaborator
Our laboratory investigates the molecular mechanisms by which transmembrane proteins (referred to as “cargo”) are sorted to different compartments of the endomembrane system in eukaryotic cells. The system comprises an array of membrane-enclosed organelles including the endoplasmic reticulum (ER), the Golgi apparatus, the trans-Golgi network (TGN), endosomes, lysosomes, lysosome-related organelles (LROs, e.g., melanosomes), and various domains of the plasma membrane in polarized cells, such as epithelial cells and neurons (Figure 1). Transport of cargo between these compartments is mediated by vesicular/tubular carriers that bud from a donor compartment, translocate through the cytoplasm, and eventually fuse with an acceptor compartment. Work in our laboratory focuses on the molecular machineries that mediate these processes, including (1) sorting signals and adaptor proteins that select cargo proteins for packaging into the transport carriers, (2) microtubule (MT) motors and organelle adaptors that drive movement of the transport carriers and other organelles through the cytoplasm, and (3) tethering factors that promote fusion of the transport carriers to acceptor compartments. We study the machineries in the context of various intracellular transport pathways, including endocytosis, recycling to the plasma membrane, retrograde transport from endosomes to the TGN, biogenesis of lysosomes and LROs, autophagy, and polarized sorting in epithelial cells and neurons. We apply knowledge gained from this basic research to the elucidation of disease mechanisms, including congenital disorders of protein traffic, such as the pigmentation and bleeding disorder Hermansky-Pudlak syndrome (HPS), hereditary spastic paraplegias (HSPs), and other neurodevelopmental disorders.
Figure 1.
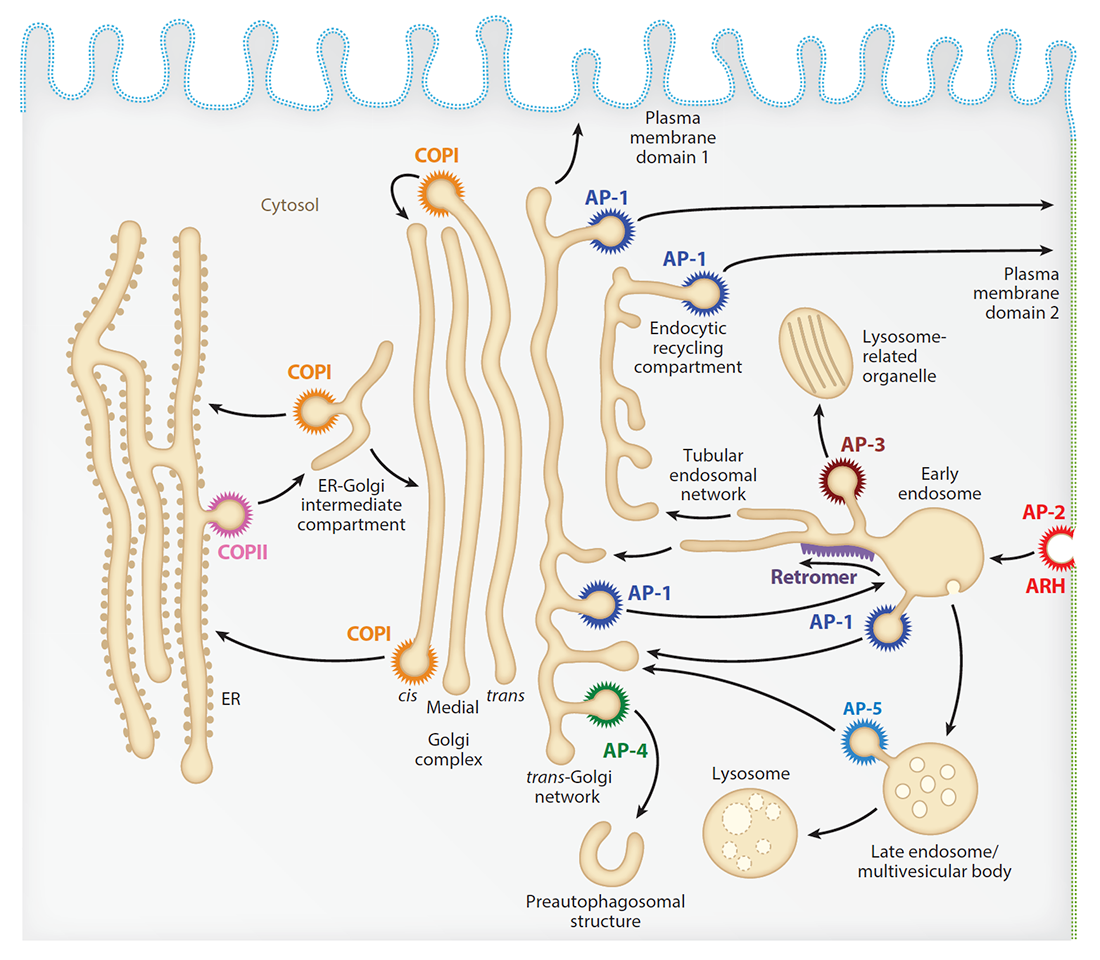
Click image to view.
Schematic representation of the endomembrane system of eukaryotic cells showing the localization of coats involved in protein sorting
ARL8 relieves SKIP autoinhibition to enable coupling of lysosomes to kinesin-1.
Long-range movement of organelles relies on coupling to microtubule motors, a process that is often mediated by adaptor proteins. In many cases, the coupling involves organelle- or adaptor-induced activation of the microtubule motors by conformational reversal of an auto-inhibited state. This past year, we discovered that a similar regulatory mechanism operates for an adaptor protein named SKIP (also known as PLEKHM2). SKIP binds to the small GTPase ARL8 on the lysosomal membrane to couple lysosomes to the anterograde microtubule motor kinesin-1. Structure-function analyses of SKIP revealed that the C-terminal region, comprising three pleckstrin homology (PH) domains, interacts with the N-terminal region, comprising ARL8– and kinesin-1–binding sites. The interaction inhibits coupling of lysosomes to kinesin-1 and, consequently, lysosome movement toward the cell periphery. We also found that ARL8 not only recruits SKIP to the lysosomal membrane, but also relieves SKIP auto-inhibition, promoting kinesin-1–driven, anterograde lysosome transport. The findings demonstrate that SKIP is not merely a passive connector of lysosome-bound ARL8 to kinesin-1 but is itself subject to both intra- and inter-molecular interactions that regulate its function.
SNX19 restricts endolysosome motility through contacts with the endoplasmic reticulum.
In addition to coupling to microtubule motors, interactions with other organelles also regulate the movement of endolysosomes within the cytoplasm. This past year, we found that the sorting nexin protein SNX19 tethers endolysosomes to the ER, reducing their motility and contributing to their concentration in the perinuclear area of the cell. Tethering depends on two N-terminal transmembrane domains that anchor SNX19 to the ER, and a Phox homology domain (PX) domain that binds to phosphatidylinositol 3-phosphate on the endolysosomal membrane. Two other domains named PXA and PXC negatively regulate the interaction of SNX19 with endolysosomes. The positioning and movement of endolysosomes within the cell are thus the result of a balance between movement driven by microtubule motors and immobilization by tethering to the ER.
Structure of human ATG9A, the only transmembrane protein of the core autophagy machinery
We also continued our studies on autophagy. A major achievement was the resolution of the atomic structure of the transmembrane autophagy protein ATG9A. In collaboration with the groups of Anirban Banerjee, Jiansen Jiang, and José Faraldo-Gomez, we succeeded in obtaining a 2.9-Ångstrom resolution cryo–EM structure of human ATG9A. The structure revealed a novel fold with a homotrimeric domain-swapped architecture, several membrane spans, and a network of branched cavities, consistent with ATG9A being a transmembrane lipid transporter. In addition, structure-guided molecular simulations predicted that ATG9A causes membrane bending, explaining the localization of this protein to small vesicles and highly curved edges of growing autophagosomes.
ATG9A transport to the cell periphery by RUSC2–mediated coupling to kinesin-1
ATG9A cycles between the TGN in the perinuclear area and pre-autophagosomal structures in the peripheral area of the cell. In previous work, we showed that export of ATG9A from the TGN into transport vesicles is mediated by the adaptor protein 4 (AP-4) complex. We recently found that the AP-4 accessory protein RUSC2 couples ATG9A–containing vesicles to the plus-end-directed microtubule motor kinesin-1 via an interaction between a disordered region of RUSC2 and the kinesin-1 light chain (KLC). The interaction is counteracted by the microtubule-associated WD40–repeat domain 47 protein (WDR47). The findings uncovered a mechanism for the peripheral distribution of ATG9A–containing vesicles, involving the function of RUSC2 as a kinesin-1 adaptor and WDR47 as a negative regulator of this function.
ATG9A enables lipid mobilization from lipid droplets.
Further work on ATG9A showed that depletion of the protein not only inhibits autophagy but also increases the size and/or number of lipid droplets in human cell lines and in C. elegans. Moreover, ATG9A depletion blocks transfer of fatty acids from lipid droplets to mitochondria and, consequently, utilization of fatty acids in mitochondrial respiration. ATG9A localizes to vesicular-tubular clusters (VTCs) that are tightly associated with an ER subdomain enriched in another transmembrane protein, TMEM41B, and are also in close proximity to phagophores, lipid droplets, and mitochondria. The findings indicate that ATG9A plays a critical role in lipid mobilization from lipid droplets to autophagosomes and mitochondria, highlighting the importance of ATG9A in both autophagic and non-autophagic processes.
Regulation of LC3B levels by ubiquitination and proteasomal degradation
In addition to working on ATG9A structure and trafficking, we investigated the mechanisms of autophagy regulation. To this end, we conducted a genome-wide CRISPR-Cas9 knockout screen using cells expressing endogenous LC3B (a microtubule-associated protein that is central to the autophagy pathway) tagged with GFP–mCherry as a reporter, an approach that allowed us to identify the ubiquitin-activating enzyme UBA6 and the hybrid ubiquitin-conjugating enzyme/ubiquitin ligase BIRC6 as novel autophagy regulators. We found that the enzymes cooperate to mediate mono-ubiquitination and proteasomal degradation of LC3B, thus limiting the pool of LC3B available for autophagy. Depletion of UBA6 or BIRC6 raised the level of cytosolic LC3B, enhancing the degradation of autophagy adaptors and the clearance of intracellular proteins aggregates. The finding could be the basis for the development of pharmacological inhibitors of UBA6 or BIRC6 for the treatment of protein-aggregation disorders.
The Golgi-associated retrograde protein (GARP) complex is critical for maintenance of the Golgi glycosylation machinery.
The Golgi apparatus is a central hub for intracellular protein trafficking and glycosylation. This past year and in collaboration with the group of Vladimir Lupashin, we found that the Golgi-associated retrograde protein (GARP) complex is critical for the maintenance of the Golgi glycosylation machinery in the Golgi apparatus. We observed that depletion of GARP subunits impairs the modification of N- and O-glycans and reduces the stability of glycoproteins and Golgi enzymes. Moreover, GARP–knockout (KO) cells exhibit reduced retention of glycosylation enzymes in the Golgi apparatus and their mis-sorting to the endolysosomal system. The findings led us to propose that the endosomal system is part of the trafficking itinerary of Golgi enzymes and that the GARP complex is essential for recycling and stabilization of the Golgi glycosylation machinery.
Publications
- Guardia CM, Tan XF, Lian T, Rana MS, Zhou W, Christenson ET, Lowry AJ, Faraldo-Gómez JD, Bonifacino JS, Jiang J, Banerjee A. Structure of human ATG9A, the only transmembrane protein of the core autophagy machinery. Cell Rep 2020;31:107837.
- Keren-Kaplan T, Bonifacino JS. ARL8 relieves SKIP autoinhibition to enable coupling of lysosomes to kinesin-1. Curr Biol 2020;31:540-554.
- Guardia CM, Jain A, Mattera R, Friefeld A, Li Y, Bonifacino JS. RUSC2 and WDR47 oppositely regulate kinesin-1-dependent distribution of ATG9A to the cell periphery. Mol Biol Cell 2021;32(21):ar25.
- Saric A, Freeman SA, Williamson CD, Jarnik M, Guardia CM, Fernandopulle MS, Gershlick DC, Bonifacino JS. SNX19 restricts endolysosome motility through contacts with the endoplasmic reticulum. Nat Commun 2021;12:4552.
- Mailler E, Guardia CM, Bai X, Jarnik M, Williamson CM, Li Y, Maio M, Golden A, Bonifacino JS. The autophagy protein ATG9A enables lipid mobilization from lipid droplets. Nat Commun 2021;12:6750.
Collaborators
- Anirban Banerjee, PhD, Unit on Structural and Chemical Biology of Membrane Proteins, NICHD, Bethesda, MD
- Mark Cookson, PhD, Laboratory of Neurogenetics, NIA, Bethesda, MD
- José Faraldo-Gómez, PhD, Theoretical Molecular Biophysics Laboratory, NHLBI, Bethesda, MD
- Spencer Freeman, PhD, Hospital for Sick Children, Toronto, Canada
- Andy Golden, PhD, Laboratory of Biochemistry and Genetics, NIDDK, Bethesda, MD
- Jiansen Jiang, PhD, Laboratory of Membrane Proteins and Structural Biology, NHLBI, Bethesda, MD
- Yan Li, PhD, Protein/Peptide Sequencing Facility, NINDS, Bethesda, MD
- Vladimir Lupashin, PhD, University of Arkansas for Medical Sciences, Little Rock, AR
- Forbes D. Porter, MD, PhD, Section on Molecular Dysmorphology, NICHD, Bethesda, MD
- Anand Swaroop, PhD, Neurobiology Neurodegeneration & Repair Laboratory, NEI, Bethesda, MD
- Chiara Zurzolo, PhD, Pasteur Institute, Paris, France
Contact
For more information, email bonifacinoj@helix.nih.gov or visit https://www.nichd.nih.gov/research/atNICHD/Investigators/bonifacino.