Physiological, Biochemical, and Molecular-Genetic Events Governing the Recognition and Resolution of RNA/DNA Hybrids

- Robert J. Crouch, PhD, Head, Section on Formation of RNA
- Susana M. Cerritelli, PhD, Staff Scientist
- Kiran Sakhuja, MS, MSc, Biologist
- Caitlin Darling, BS, Postbaccalaurate Fellow
- Emily Helm, BA, BS, Postbaccalaureate Fellow
- Matthew Roy, BS, Postbaccalaurate Fellow
- Xiang Zheng, BS, Postbaccalaureate Fellow
Damaged DNA is a leading cause of many human diseases and disorders. We study the formation and resolution of RNA/DNA hybrids, which occur during DNA and RNA synthesis. Such hybrid molecules may lead to increased DNA damage but may also play critical roles in normal cellular processes. We are interested in how RNA/DNA hybrids are resolved and in the role that ribonucleases H (RNases H) play in their elimination. Two classes of RNases H, Class I and Class II, are present in most organisms.
Patients with mutations in RNASEH1 exhibit a typical mitochondrial myopathy phenotype (muscular). Our studies were the first to show that RNase H1 is essential for the maintenance of mitochondrial DNA. Mice deleted for the Rnaseh1 gene arrest embryonic development at day 8.5 as a result of failure to amplify mitochondrial DNA. Aicardi-Goutières syndrome (AGS), a severe neurological disorder with symptoms appearing at or soon after birth, can be caused by defective human RNase H2. We are examining mouse models of AGS to gain insight into the human disorder. To understand the mechanisms, functions, substrates, and basic molecular genetics of RNases H, we employ molecular-genetic and biochemical tools in yeast and mouse models.
Differences between Class I and Class II RNases H
Many of our investigations over the years focused on RNase H1. RNase H1 recognizes the 2′-OH of four consecutive ribonucleotides, while the DNA strand is distorted to fit into a pocket of the enzyme. Thus, the enzyme requires more than one ribonucleotide for cleavage of RNA in RNA/DNA hybrids. In both eukaryotes and prokaryotes, RNases H1 consist of a single polypeptide. In contrast, RNase H2 is a complex of three distinct polypeptides in eukaryotes but a single polypeptide in prokaryotes. The catalytic subunit of the hetero-trimeric RNase H2 of eukaryotes is similar in its primary amino-acid sequence to the prokaryotic enzyme. RNase H2 can recognize and cleave both RNA/DNA hybrids and a single ribonucleotide or the transition from the ribonucleotide in the case of RNA–primed DNA synthesis (e.g., rrrrrDDDD in DNA—italics indicate transition from ribonucleotide to deoxyribonucleotide) [References 1 & 2].
Several types of RNA/DNA hybrid structure are formed, and they are processed differently. Simple RNA/DNA hybrids consist of one strand of RNA paired with one strand of DNA. The HIV–AIDS reverse transcriptase (RT) forms such hybrids when copying its genomic RNA into DNA. The RT also has an RNase H domain that is structurally and functionally similar to the class I cellular RNase H and is necessary for several steps of viral DNA synthesis. R-loop hybrids (three-stranded nucleic acid structures) have two separated DNA strands, with one hybridized to RNA while the other is in a single-stranded form. Such structures sometimes form during transcription and can lead to chromosomal breakage. However, they are also part of the normal process of switching (recombination) from one form of immunoglobulin to another, resulting in distinct isoforms of antibodies. Another form of hybrid are single or multiple ribonucleotides incorporated into DNA during replication [Reference 1]. The first two types of hybrid are substrates for class I and II RNases H. The third is uniquely recognized by type 2 RNases H.
Dual activities of RNase H2; Aicardi-Goutières syndrome
Eukaryotic RNases H2 recognize and resolve RNA hybridized or covalently attached to DNA (two chemically distinct structures) using the same catalytic mechanism for hydrolysis. RNase H2 mutations that reduce catalytic activity, or fail to properly interact with in vivo substrates, cause Aicardi-Goutières syndrome (AGS). Mutations in seven genes are known to cause AGS, with more than 50% of AGS patients having mutations in any of the three subunits of RNase H2. We previously expressed (in Escherichia coli) and purified human RNases H2 with mutations corresponding to several of those seen in AGS patients; one such mutation, RNASEH2A–G37S (G37S), has significant loss of RNase H2 activity. Using the 3D structure of the human enzyme that we had determined, we could locate all known mutations in RNase H2 that cause AGS. The wide distribution of the mutations suggests that modest changes in stability and interaction with other unknown proteins, as well as loss of catalysis, can all cause AGS. A mutation near the catalytic center of G37S found in some AGS patients results in low RNase H2 activity for both embedded ribonucleotides in DNA and RNA/DNA hybrids [Reference 1]. We are developing mouse models of AGS to clarify which defects are associated with each RNase H2 activity.
Mice bearing the G37S mutation in homozygous form are perinatal lethal, i.e., either dead at birth or die within a few hours of birth [Reference 1]. Mutations in another gene, TREX1 (which encodes a nuclease), also cause AGS, and it has been shown that homozygous knockout (KO) mice are viable but die after a few weeks owing to a cardiomyopathy that can be prevented by blocking either an innate or adaptive immune response. In contrast, the G37S–mutant perinatal lethality and the fact that RNase H2 KO mice die during early embryogenesis suggest a more severe defect than that seen in TREX1–KO mice. Damaged DNA that finds its way into the cytoplasm can be sensed by the cGAS protein producing the small molecule cGAMP, which interacts with the Sting protein, an important protein for the DNA–sensing in the innate immune pathway. Mice that are homozygous for G37S and deleted for the cGAS or Sting genes are mostly perinatal lethal but no longer exhibit increases in ISGs (interferon-stimulated genes). Interestingly, a small fraction of the double G37S–Sting KO are viable, indicating only limited involvement of ISGs in perinatal lethality. Further studies are under way, which we expect will lead us to the cause of lethality.
To distinguish among the defects that persistent RNA/DNA hybrids and single ribonucleotides joined to DNA cause in vivo, Hyongi Chon, a former postdoctoral fellow, rationally designed a modified RNase H2 to make an enzyme unable to cleave single ribonucleotides embedded in DNA but that retained RNA/DNA hydrolytic activity. The mutant enzyme, which we call RED (ribonucleotide-excision deficient), resolves RNA/DNA hybrids, which are substrates of both RNase H1 and RNase H2. Unlike the mouse and human RNases H2, RNase H2 activity is not required in the yeast Saccharomyces cerevisiae. Employing the ease of genetic mutation studies in yeast, we demonstrated that in yeast producing the RNase H2RED the enzyme acted in vivo by leaving embedded ribonucleotides (rNMPs) in DNA but was potent in removing RNA in RNA/DNA hybrids.
Embryonic lethality of mice Rnaseh2b–KO strains has been attributed to accumulation of rNMPs in DNA, but lethality could be the result of loss of RNA/DNA hydrolysis or a combination of both rNMP and RNA/DNA hydrolysis defects [References 1 & 2]. To distinguish among the possible causes of embryonic lethality, we generated a mouse that produces the RNase H2RED enzyme. Mouse embryonic fibroblasts (MEFs) derived from Rnaseh2RED mice have the same high level of rNMPs as seen in Rnaseh2b–KO MEFs [Reference 2]. Interestingly, the Rnaseh2RED mice die around the same time as the Rnaseh2b–KO mice. Therefore, lethality of the KO and RED RNase H2 mouse strains may be caused by increased rNMPs in genomic DNA. Rnaseh2aG37S/RED embryos also arrest at approximately the same stage as Rnaseh2aRED/RED embryos because of better association of RNase H2RED than RNase H2G37S with DNA substrate containing embedded rNMPs. The result is important because some RNase H2–AGS patients have similar compound heterozygous mutations in which there may be a dominant mutated enzyme.
Our studies on RNase H2-RED have permitted us and others to assign specific substrates to each of the two activities and determine which functions are related to various phenotypes seen when RNase H2 is absent [References 1–3]. One of our goals is to produce an RNase H2 with robust incision at single rNMPs in DNA but with poor RNA/DNA hybrid cleavage (hybrid-defective [HD]) to complement the RNase H2-RED enzyme. We have identified amino-acid changes that appear to produce such RNase H2-HD and are in the early stages of studies in both cell cultures and mice.
Abasic substrates
Incorporated rNMPs embedded in DNA could be converted to abasic sites in which the flanking dNMPs would be connected by a ribose phosphate rather than an rNMP. In collaboration with the Storici and Tell groups, we examined the abilities of eukaryotic RNases H2 to cleave substrates containing a single ribose abasic (only sugar phosphate) site in duplex DNA. Prokaryotic RNases HII, but not eukaryotic RNases H2, can recognize and cleave at rNMPs in duplex DNA. Little was known about abasic sites in RNA until, in collaboration with Vivian Cheung’s group, we discovered that abasic sites are present in RNAs of yeast and human cells, and likely in all organisms. The abasic sites are located in or near R-loops. The Cheung group had previously shown that MPG (N-methylpurine DNA glycosylase) and APE1 (apurinic/apyrimidinic endonuclease 1) interact with R-loops. MPG is a glycosylase that removes the base of DNA, and, as reported in our papers with Cheung, or RNA, which in turn can be cleaved by the APE1. Abasic sites in DNA are repaired by Ape1 excision using the complementary DNA strand as template for repair. RNAs have no template to correct for the absence of a base. The association of abasic sites with R-loops is intriguing and suggests that RNAs complexed with DNA are protected from RNase H activities, similar to stable R-loops present at transcription start sites of highly transcribed genes. In theory, the DNA of R-loops could serve as templates for repair of the abasic site, in which case the abasic site would survive longer than abasic sites in single-stranded RNA, giving the appearance of association of abasic sites with R-loops. However, binding of MPG and APE1 suggests that R-loops might be good substrates for MPG to generate the abasic sites.
Loss of RNase H1 in early B cell development activates the mitochondrial unfolded protein response without affecting the nuclear R-loops.
We made a knockout of the mouse Rnaseh1 gene and discovered that two proteins of RNase H1 are produced from a single mRNA by a leaky scanning method for differential translation. One protein is localized to the nucleus and a second is targeted to mitochondria. Nuclear DNA replication begins at fertilization, with mitochondrial DNA (mtDNA) beginning amplification several days later. We observed early embryonic death shortly after mtDNA replication should have begun, thereby linking absence of mtDNA replication with death. We were curious to see the contribution effects in a system less complicated then embryonic development. We chose mouse B cell development because (1) B cells are not required for viability when mice are housed in a germ-free environment; (2) B cell development occurs in only a few rounds of cell duplication; (3) resting B cells are in G0, providing a population of cells that respond together when stimulated; (4) many useful tools for analyses and manipulation are available; and (5) RNase H has potential known substrates in B cell development.
We generated an Rnaseh1 conditional KO mouse strain in which we can specifically knocked out the gene using a CRE-lox method with the Mb1 (the Mb1 gene encodes the Ig-α signaling subunit of the B cell antigen receptor) promoter–driving CRE. Transcripts of Mb1 are initiated from the earliest stage of B cell and persist until plasmacytes are formed. B cells develop to the resting stage, at which point they can be stimulated to undergo isotype switching by class switch recombination (CSR), ultimately producing circulating antibodies. We found that Mb1-CRE KO of the Rnaseh1 gene resulted in little or no circulating antibodies but did produce resting B cells, although yielding half as many B cells as did wild-type (WT) mice. Stimulation of these B cells initiated transitioning from G0 to G1 phase of the cell cycle, but essentially they never entered S-phase. The resting B cells had no intact Rnaseh1 gene, no mtDNA, the cells had no RNase H1 activity, and mitochondria exhibited abnormal morphology. We performed RNA-Seq analyses of resting and 24 h–stimulated mutant and WT B cells to discover genes related to loss of mtDNA and/or a nuclear DNA–damage response. Pathways that exhibited reductions were the cell-cycle, the immune system, DNA replication, mitochondrion, RNA processing, and ribosomes. The 50% yield of resting B cell in the KO strain must occur during cell amplification in bone marrow. The loss of RNase H1 was initiated just prior to cell amplification and might limit the number of cell cycles. It is also possible that defects affecting the time of residence of the B cells in the bone marrow niche are affected. Loss of the Nidogen1 gene (NID1) results in reduction of resting B cells to 50% of normal, the same as in our KO mice. We noticed a significant difference between WT and mutant resting B cells for the NID1 transcripts.
The list of genes with the highest fold difference between resting and stimulated KO mice are Atf5, Gdf15, Atf3, Hspa9, and Ddit3. Atf5, Atf3, and Ddit3 are all hallmarks of the unfolded mitochondrial response (UPRmt). The activation of the UPRmt indicates that loss of mtDNA takes precedence over nuclear DNA damage response, just as we observed in embryonic development when the Rnaseh1 gene was deleted in the male and female gametes. We checked the presence of R-loops by DRIP-Seq and, surprisingly, found no alteration in R-loops indicating the lack of RNase H1 in processing these structures.
Ataxic mouse model of lissencephaly-5
R-loops are three-stranded structures in which RNA loops out a DNA strand by annealing to the non-displaced DNA strand. It has been proposed that triplet expansion disease could result from improper realigning when the RNA is removed. We were particularly interested to see a mouse with severe ataxia appear in a mating with mice bearing mutations in the Rnaseh1 gene and wished to understand the origin of the phenotype in this mouse. A point mutation in the Lamb1 gene (which encodes the basement-membrane protein laminin subunit β1) rather than a triplet-expansion was discovered as the cause for the cerebellar ataxia. Mutations in the LAMB1 gene of human patients are known causes of lissencephaly-5, a neuronal migration disorder with characteristic protrusions of neurons. Lamb1 is a part of a complex of three similar proteins, a1, b1, and c1, that interact with dystroglycans and are part of congenital muscular dystrophies. One model for assembly of the complexes (Figure 1, right-hand panel) posits that these basement-membrane proteins interact in an umbrella-like manner with the C-terminal coiled-coiled regions intertwined (blue) and the N-terminal domain of each protein interacting with one or more adjacent a1 (red), b1(green), c1 (orange) complexes forming an external canopy. The coiled-coiled region forms a handle of the umbrella [McKee KK, et al., Matrix Biology 2021;98:49-63]. The mutation we found in Lamb1 is in the N-terminal loop of Lamb1, where an exposed serine has been substituted with a leucine. Structural studies could be used to support a role for the ser in protein-protein interactions. We performed multiple H&E staining and immunofluorescent studies (Figure 1, left-hand panel), which showed abnormal cerebellar lobe folding when the Lamb1 Ser-Leu mutation was homozygous but not when heterozygous. Lobes VI and VII were the most disordered, with an unusual area, possibly associated with the fastigial nucleus and/or poorly migrated components of lobes VI and VII. Reduced Lamb1 immunofluorescence signal was seen throughout the cerebellar sections in both Lamb1 heterologous and homozygous mutants. The structural integrity of the basement membrane and a normal gate can be maintained with what appears to be 50% of normal a1, b1, c1 laminin. We expect that this interesting mouse will become a model for human lissencephaly-5.
Figure 1. Ser248Leu in mouse Lamb1
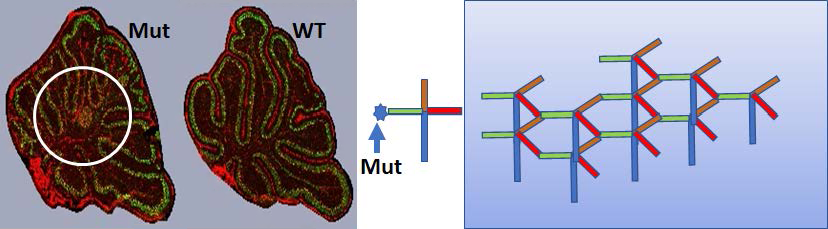
Click image to view.
Day P7 Cerebellum sections stained with Calbindin (green) and anti-Lamb1 (red). Immunofluorescent images: left Lamb1 homo; right WT. The circle highlights clear mobility defects in the mutant mouse’s cerebellum in addition to the abnormal lobe morphology. The cartoon is described in the text.
Increased incorporation of ribonucleotides in yeast DNA leads to genomic instability in the absence of RNases H.
The cellular concentration of deoxyribonucleoside triphosphates (dNTPs), the building blocks of DNA, are tightly controlled during the cell cycle to allow the replication and repair of genomic DNA. Ribonucleotide reductase (RNR) is a key enzyme, which converts ribonucleotides to deoxyribonucleotides and regulates dNTP/rNTP ratios. Cellular levels of dNTPs are much lower than those of rNTPs (ribonucleoside tri-phosphates) but increase when cells enter S-phase to facilitate genomic DNA synthesis. In addition, DNA polymerases (Pol) have active sites that select against the bulky rNTP residues in favor of dNTPs, which have a far better fit. To increase the incorporation of rNMPs in the DNA of Saccharomyces cerevisiae, we reduced the dNTP pools by depleting Rnr1, the major catalytic subunit of RNR. We observed that when dNTP pools were reduced, RNase H1 and RNase H2 were required for viability. We suggested that, under the conditions of replicative stress created by low dNTP concentrations, R-loops that accumulate in the absence of both RNase H1 and RNase H2 could become insurmountable impediments to the progression of the replication fork, inducing fork collapse and genomic instability. We further increased rNMPs incorporation in genomic DNA by depleting Rnr1 in strains that harbor DNA Pol mutants that have higher propensity to incorporate rNMPs in genomic DNA. Under such conditions, RNase H2 became essential. In addition to R-loop processing, RNase H2 initiates the ribonucleotide excision repair (RER) process, which efficiently removes single rNMPs in genomic DNA. The lethality of the triple mutant lacking RNase H2 in the DNA Pol mutant depleted of Rnr1 could be reversed in the absence of topoisomerase (Top1). We concluded that when RER is defective, Top1 processes rNMPs in DNA in a mutagenic way, inducing genome instability. When a threshold of single genomic rNMPs is exceeded in cells with limited dNTP pools and without RER, the Top1–mediated DNA damage results in severe growth defects and lethality.
Publications
- Uehara R, Cerritelli SM, Hasin N, Sakhuja K, London M, Iranzo J, Chon H, Grinberg A, Crouch RJ. Two RNase H2 mutants with differential rNMP processing activity reveal a threshold of ribonucleotide tolerance in DNA for embryonic development. Cell Rep 2018;25:1135–1145.
- Cerritelli SM, Iranzo J, Sharma S, Chabes A, Crouch RJ, Tollervey D, El Hage A. High density of unrepaired genomic ribonucleotides leads to Topoisomerase 1-mediated severe growth defects in absence of ribonucleotide reductase. Nucleic Acids Res 2020;48:4274–4297.
- Liu Y, Rodriguez Y, Ross RL, Zhao R, Watts JA, Grunseich C, Bruzel A, Li D, Burdick JT, Prasad R, Crouch RJ, Limbach PA, Wilson SH, Cheung, VG. RNA abasic sites in yeast and human cells. Proc Natl Acad Sci USA 2020;117:20689–20695.
Collaborators
- Frederic Chedin, PhD, University of California Davis, Davis, CA
- Vivian G. Cheung, MD, University of Michigan Life Sciences Institute, Ann Arbor, MI
- Lynne Holtzclaw, Microscopy and Imaging Core, OSD, IRP, NICHD
- Yosuke Mukoyama, PhD, Laboratory of Stem Cell & Neuro-Vascular Biology, NHLBI, Bethesda, MD
- Keiko Ozato, PhD, Section on Molecular Genetics of Immunity, NICHD, Bethesda, MD
- Francesca Storici, PhD, School of Biological Sciences, Georgia Institute of Technology, Atlanta, GA
- Gianluca Tell, PhD, Università degli Studi di Udine, Udine, Italy
- Kiyoshi Yasukawa, PhD, Kyoto University, Kyoto, Japan
Contact
For more information, email crouch@helix.nih.gov or visit https://sfr.nichd.nih.gov.