Childhood Neurodegenerative Lysosomal Storage Disorders

- Anil B. Mukherjee, MD, PhD, Head, Section on Developmental Genetics
- Zhongjian (Gary) Zhang, MD, PhD, Staff Scientist
- Sondra W. Levin, MD, Adjunct Scientist
- Maria B. Bagh, PhD, Visiting Fellow
- Arnab Datta, PhD, Visiting Fellow
- Vinay Patil, PhD, Visiting Fellow
- Ashleigh Bouchelion, BS, PhD, MD Student
The Section on Developmental Genetics conducts both laboratory and clinical investigations into neurodegenerative lysosomal storage disorders (LSDs) primarily affecting children. Our current research focuses on understanding the molecular mechanism(s) of pathogenesis of a group of hereditary childhood neurodegenerative LSDs called neuronal ceroid lipofuscinoses (NCLs), commonly known as Batten disease. Mutations in at least 13 different genes underlie various types of NCLs, and the list continues to grow. Currently, there is no effective treatment for any of the NCL types. The infantile NCL (or INCL) is an autosomal recessive disease caused by mutations in the CLN1 gene, which encodes palmitoyl-protein thioesterase-1 (PPT1), a lysosomal depalmitoylating enzyme. Numerous proteins, especially in the brain, undergo palmitoylation, which is a post-translational modification of polypeptides in which a long-chain saturated fatty acid (predominantly palmitate) is attached to specific cysteine residues by thioester linkage. Palmitoylation (also called S-acylation) of proteins plays important roles in signal transduction pathways, and constitutive and controlled turnover of palmitate regulates membrane association and intracellular trafficking of signaling GTPases. Moreover, numerous receptors, transporters, and ion channels are S-acylated with diverse functional consequences, including protein-protein interactions, stability, and the regulation of assembly and trafficking. Palmitoylation is catalyzed by DHHC (Asp-His-His-Cys)-palmitoyl-acyltransferases (PATs), and the removal of palmitate (depalmitoylation) is catalyzed by palmitoyl-protein thioesterases (PPTs). Twenty three palmitoylacyl transferases are encoded in the mammalian genome; four thioesterases (depalmitoylating enzymes) have been characterized. While S-acylation regulates the function of many important proteins requiring membrane anchorage, the proteins must also be depalmitoylated for recycling or for degradation by lysosomal hydrolases. Therefore, dynamic palmitoylation (palmitoylation-depalmitoylation), essential for steady-state membrane localization and signaling by many proteins, requires the interplay of both the PATs and the PPTs. Several years ago, it was discovered that mutations in a lysosomal thioesterase palmitoyl-protein thioesterase-1 (PPT1) cause accumulation of palmitoylated proteins (constituents of ceroid) in the lysosomes, leading to the devastating neurodegenerative LSD INCL. However, the precise molecular mechanism of INCL pathogenesis remains largely unclear. Children afflicted with INCL are normal at birth, but exhibit signs of psychomotor retardation by 11 to 18 months of age. By two years of age, they are completely blind as a result of retinal degeneration and, by the age 4, manifest no brain activity and remain in a vegetative state for several more years before eventual death. These grim facts underscore the urgent need for the development of rational and effective therapeutic strategies not only for INCL but also for all NCLs. Thus, our goals are to first understand the molecular mechanism(s) of pathogenesis through innovative laboratory investigations and, second, apply the knowledge gained to develop novel therapeutic strategies for INCL and possibly for other types of Batten diseases.
In our laboratory studies on INCL, we use cultured cells from patients as well as from Ppt1–knockout (Ppt1−/−) mice, which recapitulate virtually all clinical and pathological features of the disease. During the past several years, we discovered that PPT1 deficiency causes endoplasmic reticulum (ER) and oxidative stress, which at least in part contribute to neuropathology in INCL. Moreover, we delineated a mechanism by which PPT1 deficiency may disrupt the recycling of the synaptic vesicles (SVs), causing progressive loss of the SV pool size that is required for maintaining uninterrupted neurotransmission at nerve terminals. We also developed a noninvasive method, using MRI and MRS (magnetic resonance spectroscopy), to evaluate the progression of neurodegeneration in Ppt1−/− mice. The method permits repeated evaluations of potential therapeutic agents in treated animals. In addition, in collaboration with the NEI, we are conducting studies to determine whether electro-retinography can be used to assess the progressive retinal deterioration in Ppt1−/− as well as in Ppt1–knock-in (KI) mice, which carry the most common nonsense mutation found in the U.S. INCL patient population. We also discovered that the blood-brain barrier is disrupted in Ppt1−/− mice and that this pathology is ameliorated by treatment with resveratrol, which has anti-oxidant properties. More recently, we identified and characterized a non-toxic, thioesterase-mimetic, antioxidant small molecule, N-(tert-Butyl)hydroxylamine (NtBuHA), which mediates ceroid depletion, preserves motor function and modestly extends lifespan. Such compounds are potential therapeutic targets for INCL.

Click image to enlarge.
Members of the Section on Developmental Genetics
From left to right: Maria B. Bagh, Ashleigh Bouchelion, Arnab Datta, Sondra W. Levin, Anil B. Mukherjee, Vinay Patil, and Zhongjian (Gary) Zhang.
Generation of a mouse model carrying the most common nonsense mutation in the Cln1 gene
Nonsense mutations account for 5–70% of all genetic disorders. In the U.S., nonsense mutations in the CLN1 gene (also known as the PPT1 gene) underlie more than 40% of INCL cases. To evaluate nonsense suppressors in vivo, we sought to generate a reliable mouse model of INCL carrying the most common Ppt1 nonsense mutation (c.451C→T) found in the U.S. patient population. We knocked-in (KI) the c.451C→T nonsense mutation in the Ppt1 gene in C57 embryonic stem (ES) cells using a targeting vector in which LoxP flanked the Neo cassette; the cassette was removed from targeted-ES cells by electroporating Cre. Two independently targeted ES clones were injected into blastocysts to generate syngenic C57 KI mice, obviating the necessity for extensive back-crossing. Generation of Ppt1-KI mice was confirmed by DNA sequencing, which showed the presence of c.451C→T mutation in the Ppt1 gene. The mice are viable and fertile, although they developed spasticity (a "clasping" phenotype) at a median age of six months. Autofluorescent storage materials accumulated throughout the brain regions and in visceral organs. Electron-microscopic analysis of the brain and the spleen showed granular osmiophilic deposits. Elevated neuronal apoptosis was particularly evident in cerebral cortex, and abnormal histopathological and electro-retinographic (ERG) analyses attested to striking retinal degeneration. Progressive deterioration of motor coordination and behavioral parameters continued until eventual death. Our findings show that Ppt1-KI mice reliably recapitulate the INCL phenotype, providing a platform for testing the efficacy of existing and novel nonsense-suppressors alone or in combination with other drugs in vivo.
Discovery of a pathogenic link between two of the most lethal NCLs
In multicellular organisms, the lysosome is the major degradative organelle responsible for disposing off the damaged macromolecules and organelles from the cell. It has been reported that impaired lysosomal degradative capability leads to pathogenesis of many neurodegenerative disorders including LSDs. As noted above, neurodegeneration is a devastating manifestation in the majority of the more than 50 LSDs. Moreover, lysosomal degradative capability has been reported to be impaired in several late-onset neurodegenerative diseases such as Alzheimer’s, Huntington’s, and Parkinson’s diseases. Cathepsin D (CD) is a major lysosomal aspartic protease in lysosomes. Lysosomal CD activity catalyzes degradation and clearance of exogenous as well as endogenous macromolecules and damaged organelles delivered to the lysosome. Intracellular accumulation of undegraded long-lived proteins and other macromolecules leads to the pathogenesis of many neurodegenerative disorders. Paradoxically, both CD overexpression and CD deficiency have been reported to underlie neurodegenerative diseases. However, despite intense studies, this paradox has remained unresolved until now.
NCLs (Batten disease) are the most common (1 in 12 500 births), autosomal recessive, neurodegenerative LSDs, mostly affecting children. Mutations in 13 different genes (called CLNs) underlie various types of NCLs. Among all the NCLs, the infantile NCL (INCL) and congenital NCL (CNCL) are the most devastating diseases. Although the inactivating mutations in the CLN1 gene encoding palmitoyl-protein thioesterase-1 (PPT1) cause INCL, mutations in the CLN10/Ctsd gene encoding CD underlie CNCL. We sought to determine whether there is a pathogenic link between INCL and CNCL. The synthesis of CD occurs in the endoplasmic reticulum (ER) as a pre-propeptide with a molecular mass of about 50 kDa. The cleavage of the leader peptide in the ER generates the 48 kDa precursor of mature-CD (pro-CD). In the Golgi complex, attachment of mannose 6-phosphate to pro-CD facilitates the protein's binding to endosomal/lysosomal sorting receptors. The receptor-ligand complexes then exit the trans-Golgi network in clathrin-coated intermediates and fuse with the endosomal system. The low pH of the late endosomal lumen facilitates dissociation of the receptor-ligand complexes and allows the ligand (i.e., pro-CD) to be delivered to lysosome. The pro-CD then undergoes further proteolytic cleavage by cathepsin B (CB) and cathepsin L (CL), respectively, which generates the 31 and 14 kDa fragments, non-covalent dimerization of which constitutes the mature, catalytically active CD. We used Cln1−/−/Ppt1−/− mice, which recapitulate virtually all clinical and pathological features of INCL, to test for a pathogenic link between INCL and CNCL. Our results show that despite Cln10/Ctsd overexpression, defective processing of pro-CD to mature-CD in lysosome leads to lysosomal CD deficiency causing neuropathology in INCL. Because CD deficiency underlies CNCL, we propose that CD deficiency in the lysosome is the link between INCL and CNCL. Furthermore, our results suggest that N-tert-(Butyl)hydroxylamine (NtBuHA) may have therapeutic potential for patients with INCL.
A combination of cysteamine bitartrate and N-acetylcysteine for INCL patients: a bench-to-bedside study
Previously, we discovered that both cysteamine- and N-acetylcysteine–mediated ceroid depletion appeared to counteract the pathological changes in cultured cells from INCL patients. On the basis of these results, we conducted a bench-to-bedside clinical trial to determine whether the combination of oral cysteamine bitartrate and N-acetylcysteine is beneficial for patients with NCL. Children between 6 months and 3 years of age suffering from INCL with any two of the seven most lethal PPT1 mutations were eligible for inclusion in this pilot study. All patients were recruited from physician referrals. Patients received oral cysteamine bitartrate (60 mg/kg per day) and N-acetylcysteine (60 mg/kg per day) and were assessed every 6–12 months until they had an isoelectric electroencephalogram (EEG, attesting to a vegetative state) or were too ill to travel. Patients were also assessed by electro-retinography, brain MRI and MRS, and electron-microscopic analyses of leukocytes for granular osmiophilic deposits (GRODs). Children also underwent physical and neurodevelopmental assessments on the Denver scale. Outcomes were compared with the reported natural history of INCL and that of affected older siblings. This trial was registered with ClinicalTrials.gov, number NCT00028262.
Between March 14, 2001, and June 30, 2012, we recruited ten children with INCL; one child was lost to follow-up after the first visit and nine patients (five girls and four boys) were followed for 8 to 75 months. MRI showed abnormalities similar to those in previous reports; brain volume and N-acetyl aspartic acid (NAA) decreased steadily, but no published quantitative MRI or MRS studies were available for comparison. None of the children acquired new developmental skills, and their retinal function declined progressively. Average time to isoelectric EEG (52 months, SD 13) was longer than reported previously (36 months). At the first follow-up visit, peripheral leukocytes in all nine patients showed virtually complete depletion of GRODs. Parents and physicians reported less irritability, improved alertness, or both in seven patients. No treatment-related adverse events occurred apart from mild gastrointestinal discomfort in two patients, which disappeared when liquid cysteamine bitartrate was replaced with capsules. The study was completed and the results recently published. Our findings suggest that combination therapy with cysteamine bitartrate and N-acetylcysteine is associated with delay of isoelectric EEG, depletion of GRODs, and subjective benefits, as reported by parents and physicians. Our systematic and quantitative report of the natural history of patients with INCL provides a platform for evaluating experimental therapeutic strategies in the future.
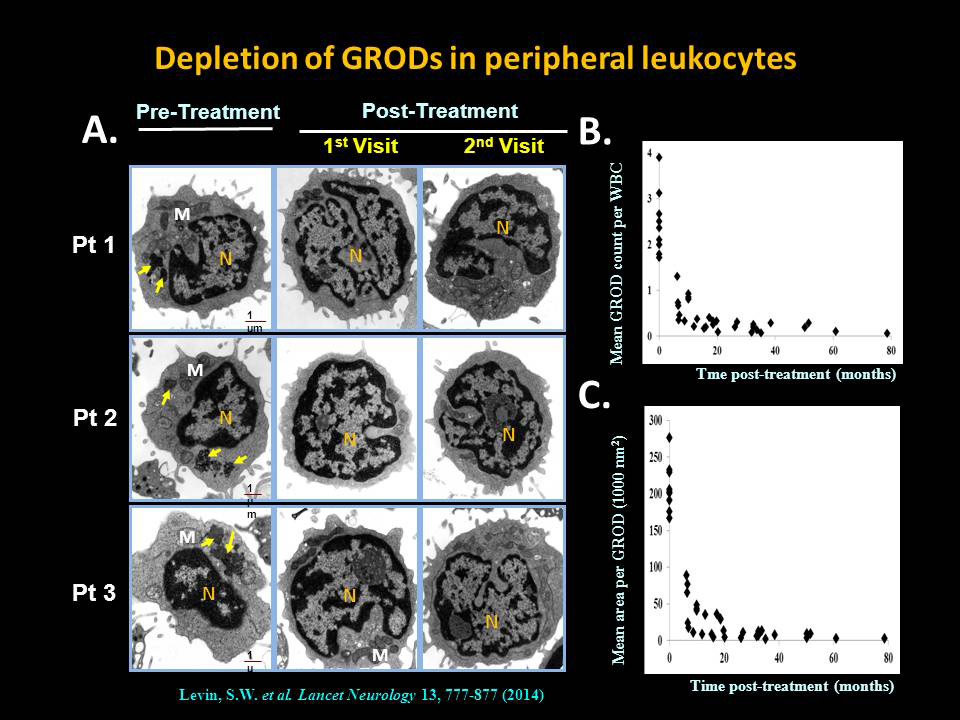
Click image to enlarge.
Depletion of lysosomal ceroid (GRODs) in INCL patients by a combination of cysteamine bitartrate and N-acetylcysteine
Oral cysteamine bitartrate and N-acetylcysteine combination mediates depletion of lysosomal ceroid (GRODs) from peripheral leukocytes of patients with INCL. Transmission electron-microscopic analyses of peripheral white blood cells from all patients were carried out at all visits, both pre-treatment and post-treatment. GRODs are irregularly shaped dark extranuclear structures that are distinct from mitochondria, secretory granules, and other membrane-enclosed cellular organelles. A. Representative micrographs from three patients are shown at three time points (pre-treatment and two post-treatment). Note that GRODs (pale green arrows) are readily detectable only in the pre-treatment image as characteristically dark, dense structures with irregular edges (clearly distinguishable from the nucleus [N] and mitochondria [M] at high magnification). B. Number of GRODs per cell and (C.) area of the GRODs for all nine patients together declined over time; the reduction began from the first visit after initiation of treatment. M=mitochondria. N=nucleus. GRODs=granular osmiophilic deposits. WBC=white blood cell.
MR spectroscopy to evaluate disease progression in INCL
As part of the pilot study to evaluate treatment benefits of cysteamine bitartrate and N-acetylcysteine, we quantitatively measured brain metabolite levels using magnetic resonance spectroscopy (MRS). A subset of two patients from a larger treatment and follow-up study underwent serial quantitative single-voxel MRS examinations of five anatomical sites. Three echo times were acquired in order to estimate metabolite T2 [quantification of the absolute concentration of metabolites using long-echo-time (TE) acquisition schemes]. Measured metabolite levels included a correction for partial volume of cerebrospinal fluid. Comparison of INCL patients was made to a reference group composed of asymptomatic and minimally symptomatic Niemann-Pick disease type C patients. In INCL patients, N-acetylaspartate (NAA) was abnormally low at all locations upon initial measurement, and further declined throughout the follow-up period. In the cerebrum (affected early in the disease course), choline and myo-inositol were initially elevated and fell during the follow-up period, whereas in the cerebellum and brainstem (affected later), choline and myo-inositol were initially normal and rose subsequently. Choline and myo-inositol levels in our patients are consistent with patterns of neuro-inflammation observed in two INCL mouse models. Low, persistently declining NAA was expected based on the progressive, irreversible nature of the disease. Progression of metabolite levels in INCL has not been previously quantified; therefore the results of this study serve as a reference for quantitative evaluation of future therapeutic interventions.
Suppression of agrin-22 production and synaptic dysfunction in Cln1−/− mice
Oxidative stress in the brain is highly prevalent in many neurodegenerative disorders including LSDs causing neurodegeneration. Despite intense studies, a precise mechanism linking oxidative stress to neuropathology in specific neurodegenerative diseases remains largely unclear. As mentioned in the introduction, INCL is a devastating neurodegenerative lysosomal storage disease caused by mutations in the CLN1 gene encoding palmitoyl-protein thioesterase-1. Previously we reported that in the brain of Cln1−/− mice, which mimic INCL, and in postmortem brain tissues from INCL patients, elevated oxidative stress is readily detectable. We used molecular, biochemical, immunohistological, and electrophysiological analyses of brain tissues of Cln1−/− mice to study the role(s) of oxidative stress in mediating neuropathology. Our results show that, via upregulation of the transcription factor CCAAT/enhancer binding protein-δ, oxidative-stress in the brains of Cln1−/− mice stimulated expression of serpin a1, which is an inhibitor of a serine protease, neurotrypsin. Moreover, in these mice suppression of neurotrypsin activity by serpin a1 inhibited the cleavage of agrin (a large proteoglycan), which substantially reduced the production of agrin-22, which is essential for synaptic homeostasis. Direct whole-cell recordings at the nerve terminals of Cln1−/− mice showed inhibition of Ca2+ currents, attesting to synaptic dysfunction. Treatment of the mice with the thioesterase-mimetic small molecule NtBuHA elevated agrin-22 levels. Our findings provide insight into a novel pathway linking oxidative stress with synaptic pathology in Cln1−/− mice and suggest that NtBuHA, which raised agrin-22 levels, may ameliorate synaptic dysfunction in this devastating neurodegenerative disease.
Publications
- Levin SW, Baker EH, Zein WM, Zhang Z, Quezado ZM, Miao N, Gropman A, Griffin KJ, Bianconi S, Chandra G, Khan OI, Caruso RC, Liu A, Mukherjee AB. Cysteamine bitartrate and N-acetylcysteine for patients with infantile neuronal ceroid lipofuscinosis: a pilot study. Lancet Neurol 2014 13:777-787.
- Bouchelion A, Zhang Z, Li Y, Qian H, Mukherjee AB. Mice homozygous for c.451C > T mutation in Cln1 gene recapitulate INCL phenotype. Ann Clin Transl Neurol 2014 1:1006-1023.
- Peng S, Xu J, Pelkey KA, Chandra G, Zhang Z, Bagh MB, Yuan X, Wu L-G, McBain CJ, Mukherjee AB. Suppression of agrin-22 production and synaptic dysfunction in Cln1−/− mice. Ann Clin Transl Neurol 2015; 2(12):1085-104.
- Chandra G, Bagh MB, Peng S, Saha A, Sarkar C, Moralle M, Zhang Z, Mukherjee AB. Cln1 gene disruption in mice reveals a common pathogenic link between two of the most lethal childhood neurodegenerative lysosomal storage disorders. Hum Mol Genet 2015 14:5416-5432.
- Baker EH, Levin SW, Zhang Z, Mukherjee AB. Evaluation of disease progression in INCL by MR spectroscopy. Ann Clin Transl Neuro 2015 2:797-809.
Collaborators
- Eva Baker, MD PhD, Radiology and Imaging Sciences, Clinical Center, NIH, Bethesda, MD
- Christopher J. McBain, PhD, Program in Developmental Neuroscience, NICHD, Bethesda, MD
- Kenneth Pelky, PhD, Program in Developmental Neuroscience, NICHD, Bethesda, MD
- Ling-Gang Wu, PhD, Synaptic Transmission Section, NINDS, Bethesda, MD
Contact
For more information, email mukherja@exchange.nih.gov or visit irp.nih.gov/pi/anil-mukherjee.