Gene Regulation in Innate Immunity

- Keiko Ozato, PhD, Head, Section on Molecular Genetics of Immunity
- Anup Dey, PhD, Biologist
- Tiyun Wu, PhD, Staff Scientist
- Mahesh Bachu, PhD, Visiting Fellow
- Chao Chen, PhD, Visiting Fellow
- Monica Gupta, PhD, Visiting Fellow
- Vishal Nehru, PhD, Visiting Fellow
- Ryota Oda, PhD, Visiting Fellow
- Sumihito Togi, PhD, Visiting Fellow
- Hiroaki Yoshii, PhD, Visiting Fellow
Macrophages and dendritic cells (DC) respond to pathogen stimuli by producing cytokines, including interferons (IFNs) and IL-1 (interleukin-1), IL-6, and TNF-alpha. While IFNs impart anti-viral and anti-microbial protection to the host, the latter cytokines are associated with inflammatory responses. IFNs are produced upon activation of IRF (interferon regulatory factor) family of transcription factors, while inflammatory cytokines are produced by the activation of NFκB. Our goal is to study the molecular pathways that direct the development and function of macrophages and DCs. To this end, we focus on the role of IRF8 in innate immunity. IRF8, a member of the IRF family, is expressed in macrophages, DCs, and microglia at high levels and is required for the production of both type I and type II IFNs. IRF8 is essential for mounting the first line of defense against various invading pathogens prior to the initiation of antigen-specific immune responses.
Transcriptionally active genes are embedded in chromatin that is dynamically exchanged, whereas silenced genes are surrounded by more stable chromatin. The chromatin environment contributes to the epigenetic states of given cells and influences transcriptional processes. We have long been working on BRD4, a chromatin-binding protein that affects transcription elongation. In this context, we are interested in histone H3.3, the variant histone that is associated with actively expressed genes. H3.3 has the unique property of being incorporated into nucleosomes and DNA only in actively transcribed genes. In contrast, H3.1, H3.2, and other standard core histones are incorporated into nucleosomes during DNA replication. For this strong association with transcription, H3.3 is implicated for epigenetic regulation. However, the process and its physiological significance of H3.3 deposition remains obscure. Our goal is to elucidate the activity of BRD4 and histone H3.3 in the context of epigenetic regulation of innate immunity.
Dynamics of histone H3.3 deposition
H3.3 is similar to the canonical histone H3 (H3.1 and H3.2) in its structure, differing only in a few amino acids. Unlike the canonical histone H3, however, H3.3 (encoded by two genes in the human and mouse) is synthesized outside of the S phase and is incorporated into chromatin along with transcription. It is thought that H3.3 associates with the preexisting canonical histone H4 to form a new nucleosome, presumably along with the histone H2AZ and the preexisting H2B. H3.3 incorporation into chromatin requires specific chromatin assembly factors distinct from those involved in replication-coupled histone deposition. HIRA is a major H3.3–specific histone chaperone dedicated to transcription-coupled H3.3 incorporation. HIRA carries the WD40 domain and B domain, through which it interacts with three other subunits to act as a chromatin assembly factor. Two other H3.3–specific chaperones, ATRX and Daxx, are reported to be involved in H3.3 incorporation into telomeres and pericentric heterochromatin.
We previously studied transcription-coupled H3.3 incorporation in IFN–stimulated genes (ISGs) in mouse embryonic fibroblasts (MEFs). Conventional ChIP analysis demonstrated that H3.3 is incorporated into ISGs upon IFN stimulation throughout the gene body and transcription end site (TES). Additional methylation of H3.3 at lysine 36 was induced after IFN stimulation, closely correlating with the H3.3 incorporation both in timing and spatial distribution. We discovered that WHSC1 (also known as NSD2) plays a significant role in H3.3 deposition and H3K36 methylation (mostly dimethylation), as evidenced by the absence of H3.3 deposition in Whsc1–/– MEFs. With additional data, we proposed a working model in which WHSC1 interacts with Brd4 and HIRA to prompt H3.3 deposition into ISGs.
IFN stimulates H3.3 deposition in macrophages: analysis of H3.3-HA knock-in mice.
To study the activity of H3.3 in vivo, we generated mouse strains in which the endogenous H3.3 genes (H3.3A, H3f3a and H3.3B, H3f3b) are replaced by HA–tagged H3.3 genes (Figure 1). We confirmed that H3.3-HA are expressed at variable levels throughout adult tissues, including all immune cells of myeloid and lymphoid lineages. We performed ChIP-seq analysis to study genome-wide distribution of H3.3A and H3.3B in bone marrow–derived macrophages from these mice. Results revealed that H3.3 deposition was rapidly induced upon IFNg stimulation, which continued for 24 h to 48 h. IFNg is a potent macrophage activator, which drives macrophages to enhance their innate immune responses. Despite the marked increase in H3.3 deposition, the total levels of H3.3A and H3.3B were not altered during this period, indicating a shift in the overall H3.3 distribution in the genome. Supporting this view, our data indicate that H3.3A and H3.3B in the intergenic regions are mobilized to support deposition in stimulated genes, a view that is supported by the reduced amounts of H3.3 in intergenic regions upon IFN stimulation.
BRD4 supports elongation of the eRNA and the coding RNA.
BRD4 has two bromodomains through which it binds to the acetylated histones H3 and H4. BRD4 also interacts with P-TEFb, the elongation factor composed of Cyclin T and CDK9. The interaction relieves the 5′-paused RNA Polymerase II (Pol II) to trigger elongation. Recent studies from various laboratories showed that BRD4 promotes cancer cell growth, including several forms of leukemia, primarily by promoting C-MYC expression. Interestingly, pharmacological inhibitors for BET bromdomains are found to be effective anti-leukemia drugs in animal models. To further study the extent of BRD4 involvement in elongation, we performed RNA-seq and ChIP-seq analyses of chromatin-bound RNA in quiescent and serum–stimulated fibroblasts. We were particularly interested in the enhancer regions that carry epigenetic histone marks, such as H3K4me1 and H3K27ac, thus affecting expression of the coding genes. We showed that serum stimulation activates transcription of many cell growth–regulated genes in their coding region as well as the enhancer (e) sequences present either in upstream or downstream. Transcription of both coding and eRNAs were dependent on BRD4’s interaction with acetylated histones, given that JQ1, a BRD4 small molecule inhibitor, blocked both RNAs. In addition, ChIP-seq data showed that BRD4 localized to the coding and enhancer regions overlapping with the localization of Pol II, supporting the view that BRD4 takes part in the elongation processes by virtue of its ability to bind to acetylated histones. To confirm the broad role of BRD4 in RNA transcription, nascent RNA was labeled with Br-UTP and immunoprecipitated by BRD4 antibody and then with BrdU antibody. Double immunoprecipitated RNA samples were analyzed by deep sequencing. We found that BRD4 is bound to newly synthesized eRNAs as well as to coding RNAs. We suggest a model wherein BRD4 directs elongation of eRNA and coding RNA (mRNA) (Figure 1). The process is guided by the distribution of acetylated histones, highlighting an epigenetic component of transcription.
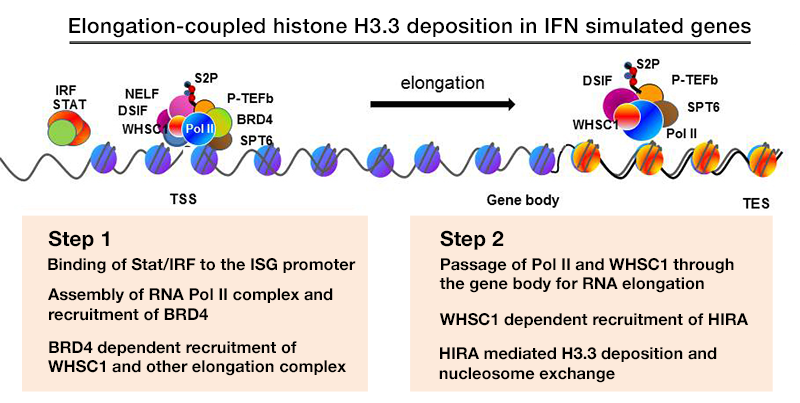
Click image to enlarge.
Figure 1. Elongation-coupled histone H3.3 deposition in IFN–stimulated genes
From left to right: Maria B. Bagh, Ashleigh Bouchelion, Arnab Datta, Sondra W. Levin, Anil B. Mukherjee, Vinay Patil, and Zhongjian (Gary) Zhang.
The role of IRF8 in autophagy and protection against infectious pathogens
Autophagy is a conserved mechanism by which misfolded self-proteins and damaged organelles (such as mitochondria) are captured and degraded through the lysosomal pathway. Macrophages and DCs activate autophagy in response to various stresses such as infection and inflammation, presumably owing to oxidative stress. Autophagy is thus a critical cellular means by which macrophages and DCs eliminate intracellular infectious agents. Autophagy has been shown to play a major role in the elimination of mycobacteria, including Mycobacterium tuberculosis, and of Salmonella. In our previous microarray analysis, we noted that a number of autophagy genes are transcriptionally induced following exposure to Toll-like receptor ligands from bacterial/viral components. However, autophagy gene induction was completely absent in macrophages and DCs from IRF8 knockout (KO) mice. Subsequent analysis found that IRF8 activates transcription of more than 15 autophagy genes that are necessary for the entire steps of autophagy in response to various stress. Autophagy genes induced by IRF8 include those for initial activation, autophagosome formation, fusion with lysosomes, and proteolytic degradation of captured materials. By quantitative ChIP assay, we found that IRF8 bound to the enhancer/promoter of many of these genes. Furthermore, retroviral transfer of Irf8 cDNA rescued expression of autophagy genes, supporting the possibility that IRF8 promotes autophagic progression in macrophages and DCs as part of innate immune responses. Thus, macrophages from IRF8–KO mice failed to form autophagosomes, as verified by diminished LC3 (light-chain 3) expression and its conversion to the membrane-associated form. Instead, IRF8–KO cells accumulated a large intracellular aggregates that contained ubiquitin-conjugated proteins and the adaptor protein SQSTM1 (p62), which are likely to be harmful to normal cellular function. This led us to study the role of IRF8 in protection against Listeria monocytogenes. Like M. tuberculosis, the bacterium resides and grows within macrophages. We showed that, upon Listeria infection, autophagy genes are induced at very high levels, which were sustained throughout infection. This was entirely absent in macrophages from IRF8–KO mice. Listeria infection also led to high IRF8 induction in wild-type macrophages. We also found that the Listeria bacilli are captured by autophagosomes in macrophages, which colocalized with LC3. However, in IRF8–KO macrophages, as a result of the paucity of autophagosomes, the bacteria multiplied rapidly without being autophagically captured. To further support the essential role for IRF8, we showed that Irf8 transfer results in partial rescue of autophagy during Listeria infection and in control of bacterial growth. Together, the results led us to propose a model in which IRF8 acts as a master regulator of autophagy and enhances host defense against invading pathogens (Figure 2).
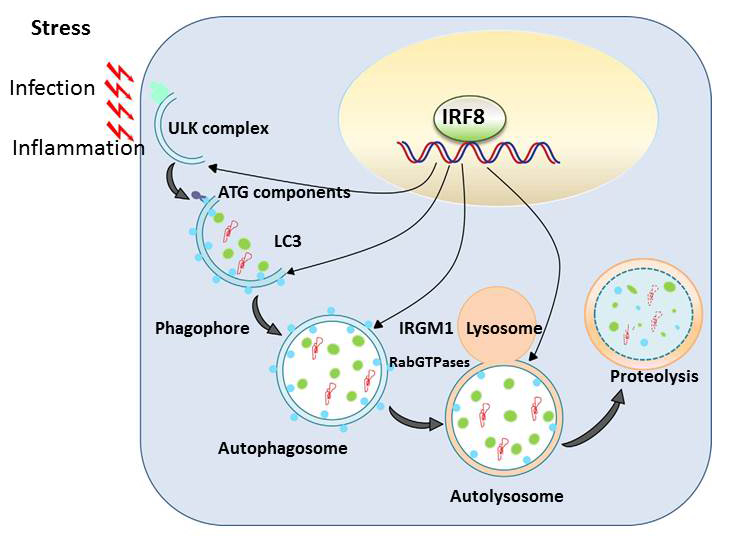
Click image to enlarge.
Figure 2. IRF8 is a master regulator of autophagy in macrophages.
Various stresses, including pathogen infection and inflammation, trigger autophagy in macrophages, activating a set of autophagy genes responsible for the entire autophagic progression. IRF8 binds to the promoter/enhancer of these genes and directs their transcription, allowing replenishment of autophagy factors. IRF8 induction of autophagy genes is required for innate clearance of intracellular bacteria, such as Listeria Monocytogenes.
In a broad view, IRF8 is viewed as a primary (pioneer) factor that sets an epigenetic landscape in the cells of innate immunity. We can thus envisage that the IRF8 gene itself assumes a chromatin signature that reflects its epigenetic status relevant to this function and lineage derivation. To this end, we mined the Encore database deposited for genome-wide chromatin marks and assembled data for various chromatin marks for the IRF4 and IRF8 loci from several human cells. The marks were then put together to show active enhancer, active transcription start site (TSS), bivalent promoter, heterochromatin and polycomb-based repression, etc. (Figure 3).
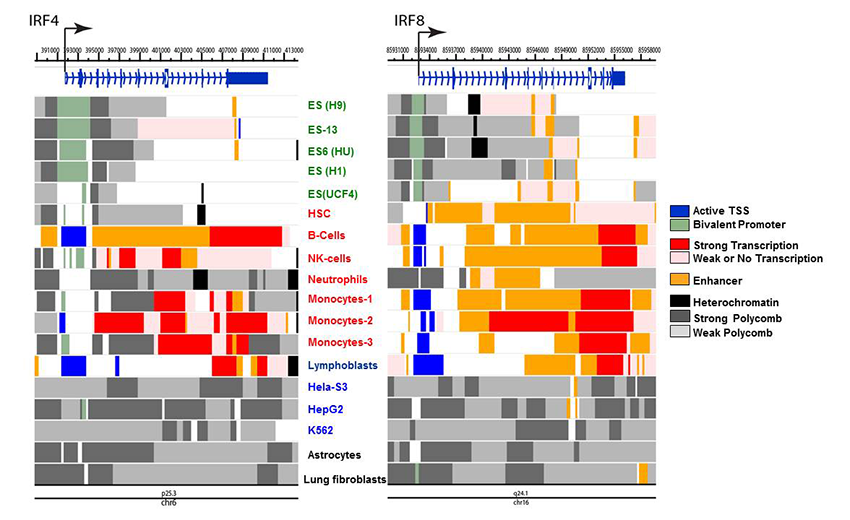
Click image to enlarge.
Figure 3. Chromatin landscape of the IRF4 and IRF8 genes in human cells
Histone modifications associated with specific epigenetic states were mined from the ENCODE database, which compiles various genome-wide analyses of human cells. Data were reorganized to visualize indicated chromatin states, using the Washington University Epigenome Browser. Cells examined were ES cells (green), primary immune cells (red), and cancer cell lines (blue). Chromatin states were assessed by the indicating modifications: Active TSS (H3K3me3, H3K4me2, H3K27Ac, H3K9Ac); Bivalent promoter (H3K4me3, and H3K27me3); Strong promoter (H3K36me3, H4K20me1, H3K79me2); Weak or no transcription (H3K36me3, H4K20me1, H3K79me2); Enhancer (H3K4me1, H3K27Ac, H3K9Ac); Heterochromatin (H3K9me3); Polycomb repression (H3K27me3).
Reflecting the similarity of the two IRF factors in their expression profiles, IRF4 and IRF8 exhibit similar chromatin marks among different cell types: in ES cells, active chromatin marks are largely absent from both genes, although IRF8 shows curious enhancer marks in all five ES cells, which are lacking in IRF4. Further, IRF8 is characterized by a strong enhancer signature in hematopoietic stem cells (HSC) and weak transcription, whereas IRF4 lacks these signatures. However, both IRF4 and IRF8 show active marks in primary B cells, NK cells, and monocytes, consistent with their expression and activities in these cells. We noted that IRF4 and IRF8 show multiple repressive marks in the cell lines HeLa S2, HepG, fibroblasts, and the myeloid lineage cells K562, and do not carry active transcription signatures. The analyses reveal previously unrecognized epigenetic marks through which IRF4 and IRF8 exert their activity in a cell type–specific manner. Similar analyses showed a distinct similarity between IRF3 and IRF7 and between IRF1 and IRF2 in their epigenetic marks.
Publications
- Kanno T, Kanno Y, LeRoy G, Campos E, Sun HW, Brooks SR, Vahedi G, Heightman TD, Garcia BA, Reinberg D, Siebenlist U, O'Shea JJ, Ozato K. BRD4 assists elongation of both coding and enhancer RNAs by interacting with acetylated histones. Nat Struct Mol Biol 2014; 12:47-57.
- Gupta M, Shin DM, Ramakrishna L, Goussetis DJ, Platanias LC, Xiong H, Morse HC 3rd, Ozato K. IRF8 directs stress-induced autophagy in macrophages and promotes clearance of Listeria monocytogenes. Nat Commun 2015; 6:6379.
- Mancino A, Termanini A, Barrozi I, Ghisletti S, Otsuni R, Prosperini E, Ozato K, Natoli G. A dual cis-regulatory code links IRF8 to constitutive and inducible gene expression in macrophages. Genes Dev 2015; 29:394-408.
- Ayithan N, Bradfute SB, Anthony SM, Stuthman KS, Bavari S, Bray M, Ozato K. Virus-like particles activate type I interferon pathways to facilitate post-exposure protection against Ebola virus infection. PLoS One 2015; 10:e0118345.
- Bradfute SB, Anthony SM, Stuthman KS, Ayithan N, Tailor P, Shaia CI, Bray M, Ozato K, Bavari S. Mechanisms of immunity in post-exposure vaccination against Ebola virus infection. PLoS One 2015; 10:e0118434.
Collaborators
- Steven B. Bradfute, PhD, United States Army Medical Institute of Infectious Diseases, Fort Detrick, MD
- Steven Holland, MD, Laboratory of Clinical Infectious Diseases, NIAID, Bethesda, MD
- Herbert Morse II, MD, Laboratory of Immunopathology, NIAID, Rockville, MD
- Dinah S. Singer, PhD, Experimental Immunology Branch, NCI, Bethesda, MD
- Tomohiko Tamura, MD, PhD, Tokyo University, Tokyo, Japan
- Jun Zhu, PhD, DNA Sequencing and Genomics Core, NHLBI, Bethesda, MD
Contact
For more information, email ozatok@mail.nih.gov or visit http://ozatolab.nichd.nih.gov.