Regulation of Childhood Growth

- Jeffrey Baron, MD, Head, Section on Growth and Development
- Kevin Barnes, PhD, Senior Research Assistant
- Julian Lui, PhD, Staff Scientist
- Youn Hee Jee, MD, Clinical Scholar
- Max Colbert, BA, Postbaccalaureate Fellow
- Melissa Jennings, BS, Postbaccalaureate Fellow
- Shanna Yue, BA, Postbaccalaureate Fellow
We investigate the cellular and molecular mechanisms governing childhood growth and development. We focus particularly on growth at the growth plate, which drives bone elongation and therefore determines height. One goal of this work is to gain insight into the many human genetic disorders that cause childhood growth failure or overgrowth. A second goal is to develop new treatments for children with severe growth disorders.
The growth plate is a thin layer of cartilage found near the ends of juvenile bones. In the growth plates new cartilage is produced through chondrocyte proliferation, hypertrophy, and cartilage matrix synthesis, and this newly formed cartilage is then remodeled into bone. The process, termed endochondral ossification, results in bone elongation, which causes children to grow in height (linear growth). Consequently, mutations in genes that regulate growth plate chondrogenesis cause abnormal bone growth and short stature in children (Reference 1). Depending on the severity and nature of the genetic abnormality, the phenotype can range from chondrodysplasias with short, malformed bones, to severe, often disproportionate, short stature, to mild proportionate short stature (Reference 1). If the genetic defect affects tissues other than the growth plate cartilage, the child may present with a more complex syndrome, which includes other clinical abnormalities (Reference 1).
Numerous gene defects that affect the growth plate and thereby cause childhood growth disorders have been identified. However, for many children who are brought to medical attention for linear growth disorders, clinical, laboratory, and genetic evaluation fail to identify the underlying etiology. Genome-wide association studies, which we helped analyze, and molecular-biological studies of growth plate biology suggest that there are hundreds of genes that control linear growth. Therefore, it is likely that many genetic causes of linear growth disorders remain to be discovered.
To discover new genetic causes of childhood growth disorders, we study families with monogenic growth disorders using powerful genetic approaches, including SNP arrays to detect large deletions, duplications, mosaicism, and uniparental disomy, combined with exome sequencing to detect single nucleotide variants and small insertions/deletions in coding regions and splice sites.
This analysis led to our identification of heterozygous mutations in the gene ACAN that cause autosomal dominant short stature with advanced bone age and premature osteoarthritis. ACAN encodes the proteoglycan aggrecan, which is an important component of cartilage extracellular matrix, including that of the growth plate. We then participated in a multi-center collaboration to study several families with this disorder to define the clinical phenotype (Reference 2). We identified 103 individuals from 20 families with heterozygous ACAN mutations. The phenotype was found to include disproportionate short stature and a history of early growth cessation. The condition was frequently associated with early-onset osteoarthritis and inter-vertebral disc disease. Most children exhibited an advanced bone age. Growth hormone therapy appeared to modestly increase growth velocity. In all subjects the disorder was inherited in an autosomal dominant fashion, thus presenting as familial short stature. However, in a subsequent study, we found that non-familial short stature can be caused by de novo ACAN mutations (Reference 3). We also recently reported that heterozygous mutations in ACAN can present with a bone age less than chronological age (Reference 3). The findings expand the known phenotypic spectrum of heterozygous ACAN mutations and indicate that this diagnosis should be considered in children without a family history of short stature and children without accelerated skeletal maturation.
Similar approaches were used to identify biallelic mutations in the BRF1 gene in a family with impaired postnatal linear growth, markedly delayed bone age, dysmorphic facies, cognitive impairment, and central nervous system anomalies (Reference 4). BRF1 encodes the RNA polymerase III transcription initiation factor 90 kDa subunit. Expression of BRF1 in yeast confirmed that the mutations affect protein function. The findings supported a previous report showing that biallelic mutations in BRF1 cause cerebellar-facial-dental syndrome. Our findings also help define the growth phenotype, indicating that the linear growth failure can become clinically evident before neurological abnormalities and that a severely delayed bone age may serve as a diagnostic clue.
In a boy with tall stature, advanced bone age, and mild dysmorphic features, exome sequencing identified a de novo missense in EZH2, a gene involved in maintaining the transcriptional repressive state of genes. The findings were consistent with previous reports that heterozygous mutations in EZH2 cause Weaver syndrome, which is characterized by tall stature, advanced bone age, characteristic facies, and variable intellectual disability. Similarly, genome-wide associated studies have implicated EZH2 in the control of height. Thus, the data indicate that EZH2 regulates skeletal growth. EZH2 encodes a histone methyltransferase that catalyzes the trimethylation of histone H3 at lysine 27 (H3K27), which serves as an epigenetic signal for chromatin condensation and transcriptional repression. To investigate the mechanisms by which this epigenetic mark affects skeletal growth, we created a mouse lacking both Ezh1 and Ezh2 in cartilage (Reference 5). The combined loss severely impaired skeletal growth. Both principal processes underlying growth plate chondrogenesis, chondrocyte proliferation and hypertrophy, were compromised. The decrease in chondrocyte proliferation was attributable in part to derepression of the cyclin-dependent kinase inhibitors Ink4a/b, while ineffective chondrocyte hypertrophy was the result of suppression of IGF signaling by the increased expression of IGF–binding proteins. Collectively, our findings reveal a critical role for H3K27 methylation in the regulation of chondrocyte proliferation and hypertrophy in the growth plate, which are the central determinants of skeletal growth.
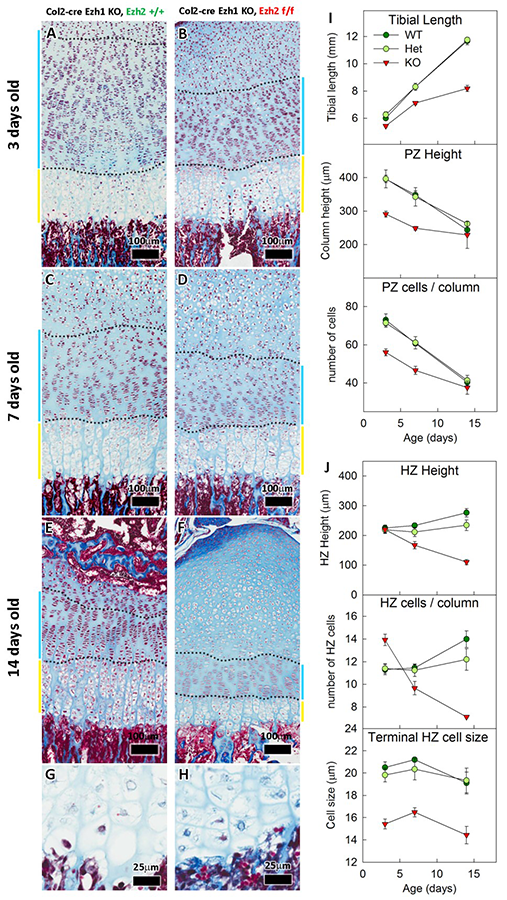
Click image to enlarge.
Figure 1. Effects of Ezh1/2 deficiency on growth plate
A-F: Histological sections of proximal tibias.
G–H: Higher magnification of hypertrophic chondrocytes at 3 days old.
I–J: Quantitative histological measurements
We also investigated the role of bone-morphogenetic proteins in the regulation of the growth plate and articular cartilage. Articular and growth plate cartilage both arise from condensations of mesenchymal cells, but ultimately develop important histological and functional differences. Each is composed of three layers: the superficial zone, the mid and deep zones of articular cartilage, and the resting, proliferative, and hypertrophic zones of growth plate cartilage. A gradient in expression of BMP–related genes has been observed across growth plate cartilage, likely playing a role in zonal differentiation. To investigate the presence of a similar expression gradient in articular cartilage, we used laser capture microdissection (LCM) to separate murine growth plate and articular cartilage from the proximal tibia into their six constituent zones and used a solution hybridization assay with color-coded probes to quantify mRNAs for 30 different BMP–related genes in each zone. In situ hybridization and immunohistochemistry were then used to confirm spatial expression patterns. We found evidence that BMP signaling gradients exist across both growth plate and articular cartilage and that the gradients contribute to the spatial differentiation of chondrocytes in the postnatal endochondral skeleton.
Additional Funding
- Clinical Center Genomic Opportunity Program (2104, ongoing): “Genetic Causes of Childhood Growth Failure”
- Merck-Serono Grant for Growth Innovation to Julian Lui (2014, ongoing): “Cartilage-Targeted Therapeutics for Growth Disorders”
- Endocrine Scholars Award in Growth Hormone Research to Julian Lui (2015, concluded): “Cartilage-Targeted IGF-I for Treatment of Growth Disorders"
- Pediatric Endocrine Society Clinical Scholar Award to Youn Hee Jee (2016, ongoing): “The role of PSD-93 in the initiation of puberty and in the etiology of pubertal delay”
- NICHD Division of Intramural Research Director’s Award Project (2016-2018, ongoing): The role of DLG2/PSD-93 in the initiation of puberty and the impact of mutations on NMDA receptor signaling and pubertal disorders
- NIH U01 award: 1U01HD086838-01A1 (2017-2021, ongoing): “Genetic Diagnosis of Childhood Growth Disorders”
Publications
- Jee YH, Baron J. The biology of stature. J Pediatr 2016 173:32-38.
- Gkourogianni A, Andrew M, Tyzinski L, Crocker M, Douglas J, Dunbar N, Fairchild J, Funari MF, Heath KE, Jorge AA, Kurtzman T, LaFranchi S, Lalani S, Lebl J, Lin Y, Los E, Newbern D, Nowak C, Olson M, Popovic J, Pruhová Š, Elblova L, Quintos JB, Segerlund E, Sentchordi L, Shinawi M, Stattin EL, Swartz J, Angel AG, Cuéllar SD, Hosono H, Sanchez-Lara PA, Hwa V, Baron J, Nilsson O, Dauber A. Clinical characterization of patients with autosomal dominant short stature due to aggrecan mutations. J Clin Endocrinol Metab 2017 102(2):460-469.
- Tatsi C, Gkourogianni A, Mohnike K, Dearment D, Witchel S, Andrade AC, Markello TC, Baron J, Nilsson O, Jee YH. Aggrecan mutations in non-familial short stature and short stature without accelerated skeletal maturation. J Endocr Soc 2017 1(8):1006–1011.
- Jee YH, Sowada N, Markello TC, Rezvani I, Borck G, Baron J. BRF1 mutations in a family with growth failure, markedly delayed bone age, and central nervous system anomalies. Clin Genet 2017 91(5):739-747.
- Lui JC, Nguyen Q, Ad M, Garrison P, Jee YH, Nilsson O, Barnes K, Baron J. Histone methyltransferases EZH1 and 2 promote skeletal growth by repressing inhibitors of chondrocyte proliferation and hypertrophy. Nat Commun 2016 7:13685.
Collaborators
- Andrew Dauber, MD, Boston Children’s Hospital, Boston, MA
- Angela Delaney, MD, Office of the Clinical Director, NICHD, Bethesda, MD
- Dimiter Dimitrov, PhD, Laboratory of Experimental Immunology, Center for Cancer Research, NCI, Frederick, MD
- Alexander A. Jorge, MD, Universidade de São Paulo, São Paulo, Brazil
- Thomas Markello, MD, PhD, Undiagnosed Diseases Program, NHGRI, Bethesda, MD
- Ola Nilsson, MD, PhD, Karolinska Institute, Stockholm, Sweden
- Katherine W. Roche, PhD, Receptor Biology Section, NINDS, Bethesda, MD
- Jan-Maarten Wit, MD, Universiteit Leiden, Leiden, The Netherlands
- Jack Yanovski, MD, PhD, Section on Growth and Obesity, NICHD, Bethesda, MD
Contact
For more information, email jbaron@mail.nih.gov or visit baron.nichd.nih.gov.