Diagnosis, Localization, Pathophysiology, and Molecular Biology of Pheochromocytoma and Paraganglioma

- Karel Pacak, MD, PhD, DSc, Head, Section on Medical Neuroendocrinology
- Thanh-Truc Huynh, MS, Biologist
- Karen T. Adams, MSc, CRNP, Research Nurse
- Tamara Prodanov, MD, Clinical Trial Database (CTDB) coordinator
- Abishek Jha, MBBS, Postdoctoral Visiting Fellow
- Liping Li, MD, PhD, Postdoctoral Visiting Fellow
- Ying Pang, MD, PHD, Postdoctoral Visiting Fellow
- Alrezk Rami, MD, PhD, Clinical Fellow
- Veronika Caisova, MS, Predoctoral Visiting Fellow
- Ivana Jochmanova, MD, Predoctoral Visiting Fellow
- Melissa Gonzales, BS, Postbaccalaureate Fellow
- Bruna Viana, BS, Postbaccalaureate Fellow
- Katherine Wolf, BS, Postbaccalaureate Fellow
- Sama Alreddawi, MD, Volunteer
- Jean Caringal, MD, Volunteer
- Garima Gupta, MD, Volunteer
- Maria Gungon, MD, Volunteer
- Amy Lopez, MD, Volunteer
- Sheila Farisha Mangelen, MD, Volunteer
- Cecilia Angela Paraiso, MD, PhD, Volunteer
- Andres Suarez Parraga, MD, Volunteer
- Valerie Valdez, MD, Volunteer
We conduct patient-oriented research into the etiology, pathophysiology, genetics, diagnosis, localization, and treatment of pheochromocytoma (PHEO) and paraganglioma (PGL), collectively abbreviated as PPGLs. PPGLs are rare, neuroendocrine tumors derived from adrenal or extra-adrenal chromaffin cells, respectively. Metastases are discovered in 3–36% of patients at the time of diagnosis. Currently, only suboptimal treatment options exist. Therefore, new therapeutic compounds targeting metastatic PPGLs are urgently needed. Our projects include both translational research—applying basic science knowledge to clinical diagnosis, pathophysiology, and treatment—and ‘reverse translation research,’ by which clinical findings lead to new concepts for pursuit by basic researchers in the laboratory. Our goals are to (1) establish new and improved methods and strategies for novel diagnostic and localization approaches to PPGL; (2) explain the molecular and cellular basis for varying clinical presentations of PPGLs and establish the pathways of tumorigenesis; (3) search for new molecular and genetic/epigenetic markers for diagnosis and treatment of metastatic PPGL; (4) introduce new therapeutic options for malignant/metastatic PPGL; and (5) facilitate new and improved collaborations and interdisciplinary studies. To achieve these goals, we enter into multidisciplinary collaborations with investigators from several NIH Institutes and outside medical centers. We link a patient-oriented component with two bench-level components. The patient-oriented component (medical neuroendocrinology) is the driving force for our hypotheses and discoveries. The two bench-level components (tumor pathogenesis/genetics and chemistry/biomarkers) emphasize, first, technologies of basic research tailored for pathway and target discovery and, second, the further development of discoveries into clinical applications.
Clinical aspects of pheochromocytoma and paraganglioma
Childhood PPGLs are often hereditary and may present with different characteristics compared with adults. Hereditary PPGLs can be separated into cluster 1 and cluster 2 tumors, which result from to mutations impacting hypoxia and kinase receptor signaling pathways, respectively. The objective was to identify differences in presentation of PPGLs between children and adults. The study included 748 patients with PPGLs, including 95 with a first presentation during childhood. Genetic testing was available in 611 patients. Other data included locations of primary tumors, presence of recurrent or metastatic disease, and plasma concentrations of metanephrines and 3-methoxytyramine. Children showed significantly higher prevalence than adults of hereditary (80.4% vs. 52.6%), extra-adrenal (66.3% vs. 35.1%), multifocal (32.6% vs. 13.5%), metastatic (49.5% vs. 29.1%), and recurrent (29.5% vs. 14.2%) PPGLs. Tumors resulting from to cluster 1 mutations were significantly more prevalent among children than among adults (76.1% vs. 39.3%), and this paralleled a significantly higher prevalence of noradrenergic tumors, characterized by relative lack of increased plasma metanephrine, in children than in adults (93.2% vs 57.3%). We concluded that the higher prevalence of hereditary, extra-adrenal, multifocal, and metastatic PPGLs in children than in adults represents interrelated features that, in part, reflect the lower age of disease presentation of noradrenergic cluster 1 than adrenergic cluster 2 tumors. The differences in disease presentation are important to consider in children at risk for PPGLs due to a known mutation or previous history of tumor.
We then performed a retrospective study of 55 patients diagnosed at or before 21 years of age with PPGLs, with analysis of data on genetic testing and multimodal imaging. Eighty percent of patients (n = 44/55) had a germline mutation. The majority were found to have mutations either in the von Hippel-Lindau tumor suppressor (VHL) gene (38%) or in the SDHB (encoding succinate dehydrogenase complex iron sulfur subunit B) gene (25%). PHEO was present in 67% (n = 37/55) of patients and was bilateral in 51% (n = 19/37). The majority of patients with bilateral PHEOs had VHL (79%). Abdominal PGLs was present in 22% (n = 12/55), head and neck PGLs in 11% (n = 6/55), and thoracic PGLs in 2 of 55 patients. For PGLs, SDHx (succinate dehydrogenase) accounted for 72% (n = 13/18) of mutations. The rate of malignancy was 16% (n = 9/55), 56% of whom had SDHB mutations. In two-thirds of patients, functional imaging identified either extra-adrenal PGLs and/or metastatic disease. We concluded that the majority of pediatric patients with PPGLs have detectable germline mutations. Therefore, we strongly suggest that all pediatric patients with PPGLs undergo genetic testing and imaging to detect extra-adrenal paragangliomas and metastatic disease to guide treatment and follow-up.
In our review paper (Reference 1), we discussed a new molecular biology–based taxonomy that has been proposed for PPGL. Data from the Cancer Genome Atlas revealed clinically relevant prognostic and predictive biomarkers and stratified PPGLs into three main clusters. Each subgroup has a distinct molecular-biochemical imaging signature. Concurrently, new methods for biochemical analysis, functional imaging, and medical therapies have also become available. The research community now strives to match the cluster biomarkers with the most superior intervention. The concept of precision medicine has been long awaited and holds great promise for improved care. We reviewed the current and future PPGL classifications with a focus on hereditary syndromes. We discussed the current strengths and shortcomings of precision medicine, and suggested a condensed manual for diagnosis and treatment of both adult and pediatric PPGL patients. We also considered the future direction of this field, with a particular focus on how advanced molecular characterization of PPGL can improve a patient’s outcome, including cures, and ultimately disease prevention.
Hereditary pheochromocytoma and paraganglioma
Succinate dehydrogenase B (SDHB)–associated PHEOs are associated with a higher risk of tumor aggressiveness and malignancy. The aim of the present study was to evaluate first the frequency of germline SDHB mutations in apparently sporadic patients with PHEO who undergo preoperative genetic testing and, second, the ability to predict pathogenic mutations. From 2012 to 2016, 82 patients underwent a PHEO surgical resection. Sixteen were operated in the context of hereditary PHEO and were excluded from analysis. Among the 66 remaining cases, 48 were pre-operatively screened for an SDHB mutation. In addition to imaging studies with specific radiopharmaceuticals (123I-MIBG or 18F-FDOPA) for exclusion of multifocality/metastases, 36 patients underwent 18F-FDG PET/CT. From the 48 genetically screened patients, genetic testing found a germline SDHB variant in two (4.2%) cases: a variant of unknown significance, exon 1, c.14T>G (p.Val5Gly), and a most likely pathogenic mutation, exon 5, c.440A>G (p.Tyr147Cys), according to in silico analysis. Structural and functional analyses of the protein predicted that the p.Tyr147Cys mutation was pathogenic. Both tumors exhibited moderate 18F-FDG PET uptake with similar uptake patterns to non–SDHB mutated PHEOs. The two patients underwent total laparoscopic adrenalectomies. Of the remaining patients, 44 underwent adrenalectomy laparoscopically and two by laparotomy. Pathology analysis of the tumors from patients bearing two germline SDHB variants revealed a typical PHEO. Ex vivo analyses (metabolomics, SDHB immunohistochemistry, and loss of heterozygosity analysis) allowed a reclassification of the two SDHB variants as probably non-pathogenic variants. The study illustrated that SDHx mutational analysis can be misleading, even if structural and functional analyses are done. Surgeons should be aware of the difficulty of classifying new SDHB variants prior to including SDHB mutation status into a tailored surgical management strategy of a patient.
In another study, we reported a comprehensive molecular characterization of PPGLs. Multi-platform integration revealed that PPGLs are driven by diverse alterations affecting multiple genes and pathways. Pathogenic germline mutations occurred in eight PPGL susceptibility genes. We identified CSDE1, a gene encoding the RNA–binding protein cold shock domain–containing protein E1, as a somatically mutated driver gene, complementing four known drivers (HRAS, encoding a GTPase that is a protein kinase activator, RET, a gene encoding a receptor tyrosine kinase, EPAS1, a gene encoding a transcription factor involved in body response to oxygen levels, and NF1, a gene encoding a negative regulator of the ras signal transduction pathway). We also discovered fusion genes in PPGLs, involving MAML3 (encoding a transcriptional coactivator for NOTCH proteins), BRAF (encoding the serine/threonine-protein kinase B-raf), NGFR (encoding a low-affinity receptor), and NF1. Integrated analysis classified PPGLs into four molecularly defined groups: a kinase signaling subtype, a pseudohypoxia subtype, a Wnt–altered subtype driven by MAML3 and CSDE1, and a cortical admixture subtype. Correlates of metastatic PPGLs included the MAML3 fusion gene. This integrated molecular characterization provides a comprehensive foundation for developing PPGL precision medicine.
In recent years, familial pPHEO with germline mutations in the MAX (MYC associated factor X) gene has been reported in a few cases. We investigated a 25-year-old patient with multiple PHEOs associated with a nonsense germline MAX mutation. Preoperative 18F-FDOPA PET/CT revealed bilateral adrenal involvement with multiple tumors. In addition, both adrenal glands were found to have diffuse or nodular adrenal medullary hyperplasia (AMH), a histopathological feature previously described as a precursor of MEN2– and SDHB–related PHEOs but not of MAX. After bilateral adrenalectomy, different paraffin-embedded and frozen samples were analyzed for allelic imbalances of the MAX gene using allelic quantification by pyrosequencing. The expression of the protein MAX was studied by immunohistochemistry. All PHEOs, but also nodular AMH, exhibited a loss of the normal allele. By contrast, the diffuse AMH did not show loss of heterozygosity. Nevertheless, immuno-histochemistry demonstrated loss of protein MAX expression in all samples including diffuse hyperplasia, suggesting a causative role of MAX mutation for both PHEOs and AMH. The case shows that both nodular and diffuse AMH belong to the spectrum of MAX–related disease. The data support the possible continuum between nodular AMH and PHEO, expanding the qualification of micro-PHEO to nodular AMH.
Imaging of pheochromocytomas and paragangliomas
Pheochromocytoma/paraganglioma (PPGL) syndromes associated with polycythemia have previously been described in association with mutations in the von Hippel-Lindau gene. Recently, mutations in the prolyl hydroxylase gene (PHD) 1 and 2 and in the hypoxia-inducible factor 2α (HIF2A) were also found to be associated with multiple and recurrent PPGL. Such patients also presented with PPGL and polycythemia, and later on, some presented with duodenal somatostatinoma. In additional patients presenting with PPGL and polycythemia, no further mutations have been discovered. Because the functional imaging signature of patients with PPGL-polycythemia syndromes is still unknown, and because these tumors (in most patients) are multiple, recurrent, and metastatic, the goal of our study was to assess the optimal imaging approach using four different PET radio-pharmaceuticals and CT/MRI in these patients. Fourteen patients (10 women, 4 men) with confirmed PPGL and polycythemia prospectively underwent imaging by: 68Ga-DOTATATE, in 13 patients; 18F-FDG, in 13 patients; 18F-fluorodihydroxyphenylalanine (18F-FDOPA), in all 14 patients; 18F-fluorodopamine (18F-FDA), in 11 patients; and CT/MRI, in all 14 patients. Detection rates of PPGL lesions were compared between all imaging studies and stratified between the underlying mutations. 18F-FDOPA and 18F-FDA PET/CT showed similar combined lesion-based detection rates of 98.7% (95% confidence interval [CI], 92.7%–99.8%) and 98.3% (95% CI, 90.9%–99.7%), respectively. The detection rates for 68Ga-DOTATATE (35.3%; 95% CI, 25.0%–47.2%), 18F-FDG (42.3; 95% CI, 29.9%–55.8%), and CT/MRI (60.3%; 95% CI, 48.8%–70.7%) were significantly lower, irrespective of the mutation status. We concluded that 18F-FDOPA and 18F-FDA are superior to 18F-FDG, 68Ga-DOTATATE, and CT/MRI and should be the radio-pharmaceuticals of choice in this rare group of patients.
From 2007 to 2015, 175 patients with non-metastatic PPGL were evaluated by 18F-FDOPA PET/CT for initial diagnosis/staging and follow-up. 18F-FDOPA PET/CT was considered as falsely negative for at least one lesion in 10/126 (8%) patients (two sporadic, six SDHD, two SDHB PPGLs). The mRNA and protein expression levels of the LAT amino-acid transporters and a subunit of LAT1, CD98hc, were evaluated in samples with different genetic backgrounds and imaging phenotypes. The qRT-PCR (quantitative real-time PCR) and immunohistochemical analyses were performed in 14 and 16 tumor samples, respectively. The SDHx–mutated samples exhibited a significant decrease in mRNA expression of LAT3 when compared with sporadic PPGLs. There was also a statistical trend toward reduced CD98hc and LAT4 levels in SDHx vs. sporadic PPGLs. No difference was observed for LAT1/LAT2 mRNA levels. LAT1 protein was expressed in 15 out of 16 (93.75%) SDHx tumors, regardless of the 18F-FDOPA positivity. LAT1 and CD98hc were co-expressed in 6/8 18F-FDOPA-negative PPGLs. In contrast, in one case with absence of LAT1/CD98hc, 18F-FDOPA uptake was positive and attributed to LAT4 expression. We conclude that down-regulation of LAT1/CD98hc cannot explain the imaging phenotype of SDHx–related PPGLs. A reduced activity of LAT1 remains the primary hypothesis possibly due to a modification of intracellular amino acid content which may reduce 18F-FDOPA uptake.
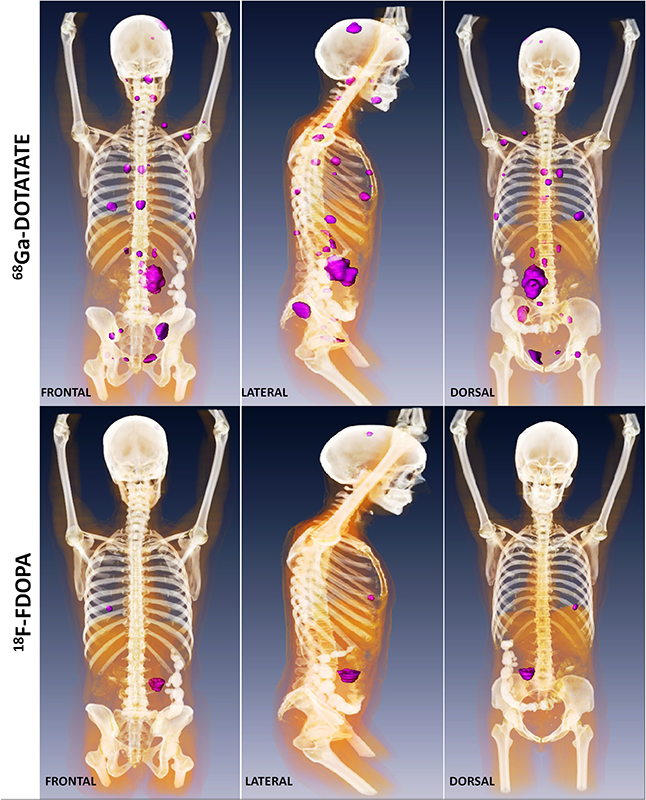
Click image to enlarge.
Figure 1. Metastatic paraganglioma detected by 68Ga-DOTATATE PET/CT and 18F-FDOPA PET/CT
Detection of metastatic paraganglioma with the novel imaging modality 68Ga-DOTATATE PET/CT compared with 18F-FDOPA PET/CT (frontal, lateral, and dorsal views)
Authors: Papadakis GZ, Bagci U, Millo CM, Janssen I, Patronas NJ, Stratakis CA, Pacak K
Therapeutic aspects of pheochromocytoma and paraganglioma
No definite cure is yet available for the neuroendocrine tumors PPGLs. Therefore, drugs with a wide spectrum of mechanisms of action are being tested to identify suitable candidates for PPGL treatment. Proteasome inhibitors have been frequently used in treating hematologic and solid tumors. They are administered individually or in combination with other regimens, to prevent severe side effects and resistance development. Because they have been shown to be efficient and are pharmaceutically available, we tested the first Food and Drug Administration–approved proteasome inhibitor bortezomib alone and in combination with another proteasome inhibitor, salinosporamid A, in PHEO cells. We showed that bortezomib induces PHEO cell death via the apoptotic pathway both in vitro and in vivo. The combination of bortezomib with salinosporamid A exhibits an additive effect on these cells and inhibits proliferation, cell migration and invasion, and angiogenesis more potently than bortezomib alone. We suggest these proteasome inhibitors, especially bortezomib, could be tested in PPGL patients, who might benefit from treatment with either the inhibitors alone or in combination with other treatment options.
Histone deacetylase inhibitors (HDACis) are a potent class of tumor-suppressive agents traditionally believed to exert their effects by loosening tightly wound chromatin, resulting in de-inhibition of various tumor suppressive genes. Recent literature however has shown altered intra-tumoral hypoxia signaling with HDACi administration not attributable to changes in chromatin structure. We sought to determine the precise mechanism of HDACi–mediated hypoxia signaling attenuation using vorinostat (SAHA), an FDA–approved class I/IIb/IV HDACi. Through an in vitro and in vivo approach utilizing cell lines for hepatocellular carcinoma (HCC), osteosarcoma (OS), and glioblastoma (GBM), we demonstrated that SAHA potently inhibits HIF-α nuclear translocation via direct acetylation of its associated chaperone, heat shock protein 90 (Hsp90). In the presence of SAHA, we found elevated levels of acetyl-Hsp90, reduced interaction between acetyl-Hsp90 and HIF-α, reduced nuclear/cytoplasmic HIF-α expression, no HIF-α association with its nuclear karyopharyin Importin, and markedly reduced HIF-α transcriptional activity. These changes were associated with downregulation of downstream hypoxia molecules such as endothelin 1, erythropoietin, glucose transporter 1, and vascular endothelial growth factor. The findings were replicated in an in vivo Hep3B HRE-Luc expressing xenograft and were associated with significant decreases in xenograft tumor size. The study highlights a novel mechanism of action of an important class of chemotherapeutic.
We investigated whether anthracyclines were able to suppress the progression of metastatic PHEO. We explored their effects on experimental mouse PHEO tumor cells, using in vitro and in vivo models, and demonstrated that anthracyclines, particularly idarubicin (IDA), suppressed hypoxia signaling by preventing the binding of hypoxia-inducible factor 1 and 2 (HIF-1 and HIF-2) to the hypoxia response element (HRE) sites on DNA. This resulted in reduced transcriptional activation of HIF target genes, including erythropoietin (EPO), phosphoglycerate kinase 1 (PGK1), endothelin 1 (EDN1), glucose transporter 1 (GLUT1), lactate dehydrogenase A (LDHA), and vascular endothelial growth factor (VEGFA), which consequently inhibited the growth of metastatic PHEO. Additionally, IDA downregulated hypoxia signaling by interfering with the transcriptional activation of HIF1A and HIF2A. Furthermore, our animal model demonstrated the dose-dependent suppressive effect of IDA on metastatic PHEO growth in vivo. Our results indicate that anthracyclines are prospective candidates for inclusion in metastatic PPGL therapy, especially in patients with gene mutations involved in the hypoxia signaling pathway.
Animal model of pheochromocytoma and cell culture studies
Approximately 30–40% of PPGLs result from germline mutations in one of the susceptibility genes, including those encoding the succinate dehydrogenase subunits A-D (SDHA–D). Up to two thirds of patients affected by SDHB mutated PPGL develop metastatic disease with no successful cure at present. For the first time, we evaluated the effects of SDHB silencing in a three dimensional (3D) culture using spheroids of a mouse PHEO cell line, in which the SDHB subunit was either silenced or not (wild-type). We investigated the role of the micro-environment on spheroid growth and migration/invasion by co-culturing SDHB–silenced or wild-type spheroids with primary cancer-activated fibroblasts (CAFs). When spheroids were co-cultured with fibroblasts, SDHB–silenced cells showed a statistically significant increase in matrigel invasion, as demonstrated by the computation of the migratory areas. Moreover, cells detaching from the SDHB–silenced spheroids moved collectively, unlike the cells of wild-type spheroids, which moved individually. Additionally, SDHB–silenced spheroids developed long filamentous formations along which clusters of cells migrated far away from the spheroid, whereas these structures were not present in wild-type spheroids. We found that lactate, largely secreted by CAFs, plays a specific role in promoting migration only of SDHB–silenced cells. Thus, in this study, we demonstrated that SDHB silencing per se increases tumor cell migration/invasion and that this micro-environment, as represented by CAFs, plays a pivotal role in enhancing collective migration/invasion in PHEO SDHB–silenced tumor cells, suggesting their role in increasing the tumor metastasizing potential.
Publications
- Crona J, Taïeb D, Pacak K. New perspectives on pheochromocytoma and paraganglioma: towards a molecular classification. Endocr Rev 2017 38(6):489-515.
- Taïeb D, Pacak K. New insights into the nuclear imaging phenotypes of cluster 1 pheochromocytoma and paraganglioma. Trends Endocrinol Metab 2017 28(11):807-817.
- Pang Y, Yang C, Schovanek J, Wang H, Bullova P, Caisova V, Gupta G, Wolf KI, Semenza GL, Zhuang Z, Pacak K. Anthracyclines suppress pheochromocytoma cell characteristics, including metastasis, through inhibition of the hypoxia signaling pathway. Oncotarget 2017 8:22313-22324.
- Jochmanova I, Wolf KI, King KS, Nambuba J, Wesley R, Martucci V, Raygada M, Adams KT, Prodanov T, Fojo AT, Lazurova I, Pacak K. SDHB-related pheochromocytoma and paraganglioma penetrance and genotype-phenotype correlations. J Cancer Res Clin Oncol 2017 143:1421-1435.
- Janssen I, Chen CC, Zhuang Z, Millo CM, Wolf KI, Ling A, Lin FI, Adams KT, Herscovitch P, Feelders RA, Fojo AT, Taieb D, Kebebew E, Pacak K. Functional imaging signature of patients presenting with polycythemia/paraganglioma syndromes. J Nucl Med 2017 58:1236-1242.
Collaborators
- Clara C. Chen, MD, Nuclear Medicine Department, Clinical Center, NIH, Bethesda, MD
- Graeme Eisenhofer, PhD, Universität Dresden, Dresden, Germany
- Abdel G. Elkahloun, PhD, Genome Technology Branch, NHGRI, NIH, Bethesda, MD
- Alessio Imperiale, MD, PhD, Hôpital de Hautepierre, Les Hôpitaux Universitaires de Strasbourg, Strasbourg, France
- Electron Kebebew, MD, Surgery Branch, NCI, Bethesda, MD
- Ron Lechan, MD, PhD, Tufts Medical Center, Boston, MA
- Jacques Lenders, MD, Radboud Universiteit, Nijmegen, The Netherlands
- W. Marston Linehan, MD, Urologic Oncology Branch, NCI, Bethesda, MD
- Alexander Ling, MD, Radiology Department, Clinical Center, NIH, Bethesda, MD
- Lani Mercado-Asis, MD, PhD, University of Santo Tomas, Manila, Philippines
- Maria J. Merino, MD, Pathology Department, NCI, Bethesda, MD
- Corina Millo, MD, PET Department, Clinical Center, NIH, Bethesda, MD
- Margarita Raygada, PhD, Section on Endocrinology and Genetics, NICHD, Bethesda, MD
- James C. Reynolds, MD, Nuclear Medicine Department, Clinical Center, NIH, Bethesda, MD
- Constantine A. Stratakis, MD, D(med)Sci, Section on Endocrinology and Genetics, NICHD, Bethesda, MD
- David Taïeb, MD, PhD, Centre de Recherche en Cancérologie de Marseille, Aix-Marseille Univerisité, Marseille, France
- Henri Timmers, MD, PhD, Radboud Universiteit, Nijmegen, The Netherlands
- Arthur S. Tischler, MD, PhD, New England Medical Center, Boston, MA
- Robert A. Wesley, PhD, Clinical Epidemiology and Biostatistics Service, Clinical Center, NIH, Bethesda, MD
- Zhengping Zhuang, MD, PhD, Surgical Neurology Branch, NINDS, Bethesda, MD
Contact
For more information, email karel@mail.nih.gov or visit http://pheopara.nichd.nih.gov.