Gene Regulation in Innate Immunity

- Keiko Ozato, PhD, Head, Section on Molecular Genetics of Immunity
- Anup Dey, PhD, Biologist
- Tiyun Wu, PhD, Staff Scientist
- Mahesh Bachu, PhD, Visiting Fellow
- Chao Chen, PhD, Visiting Fellow
- Vishal Nehru, PhD, Visiting Fellow
- Keita Saeki, MD, PhD, Visiting Fellow
- Sumihito Togi, PhD, Visiting Fellow
- Keith Sakata, BS, Postbaccalaureate Intramural Research Training Award Fellow
Macrophages and related cells recognize incoming pathogens and produce cytokines, such as interferons (IFNs) and IL-1/IL-6/TNF-alpha. While IFNs impart anti-viral and anti-microbial protection to the host, the latter cytokines are associated with inflammatory responses. IFNs are produced upon activation of the IRF (interferon regulatory factor) family of transcription factors, while inflammatory cytokines are produced by activation of the transcription factor NFκB. Our goal is to study the molecular pathways that direct the development and function of macrophages and other myeloid cells. To this end, we focus on the role of IRF8 in innate immunity. IRF8, a member of the IRF family, is expressed in macrophages, dendritic cells (DCs), and microglia at high levels and is required for the production of both type I and type II IFNs. IRF8 is essential for mounting the first line of defense against various invading pathogens prior to the initiation of antigen-specific adaptive immune responses.
Transcriptionally active genes are embedded in chromatin that is dynamically exchanged, whereas silenced genes are surrounded by more stable chromatin. The chromatin environment contributes to the epigenetic states of given cells and influences transcriptional processes. We have long been working on BRD4, a bromodomain protein that binds to acetylated histones and promotes active transcription. BRD4 is involved in the dynamic chromatin exchange that takes place in highly transcribed genes, an exchange that requires a special histone called H3.3. As a result of the association with transcription, H3.3 is implicated in epigenetic control of gene expression patterns. Our goal is to elucidate the activity of BRD4 and histone H3.3 in innate immunity.
IRF8 confers innate protection against mycobacterium infection.
IRF8 is a transcription factor important for host defense against a variety of pathogens—from viruses to bacteria. IRF8 has been reported to play a pivotal role in protection against Mycobacterium tuberculosis (TB). Recent exome sequencing efforts at NIH revealed IRF8 mutations likely associated with susceptibility to another mycobacterium, the so called non-tuberculosis mycobacterium (NTM). The majority of patients infected with NTM suffer from chronic pulmonary distress, although there are also cases of widespread, disseminated infection. Similar to TB, NTM resides and replicates within macrophages for extended periods. There are relatively few antibiotics/drugs that are effective against NTM. While BCGs have been used for a vaccine, their efficacy is not established. Moreover, the mechanism of innate immunity against mycobacteria is poorly understood. These unsolved problems pose a public health threat, as the incidence of NTM infection is rising in developed countries. In collaboration with Steven Holland and Katrin Mayer-Barber, we are investigating the role of IRF8 in the protection against NTM. As an in vitro approach, bone marrow-derived macrophages from wild type (WT) and Irf8 knockout (KO) mice were infected with NTM (M. avis complex), and bacterial growth was measured by CFU (colony-forming unit) assay after seven days of incubation. We found that bacterial counts were more than 10 times higher in Irf8 KO macrophages than in WT cells and that interferon gamma, known to facilitate innate resistance, was not effective in Irf8 KO macrophages. RNA-seq analysis found that a series of genes were induced sequentially after NTM infection in WT macrophages. Some of early genes included IL-1 and TNF, known to be important for resistance against TB. Thus, roughly one-third of 1,500 genes induced by NTM infection in WT macrophages were not induced in Irf8 KO macrophages, including several autopagy-related genes and those important for lysosomal function. Our results point to the role of autophagy lysosomal pathway in the establishment of anti-mycobacterium resistance. The idea that this pathway is critical for anti-mycobacterium innate immunity is supported first by reports that the Atg5 KO mouse, defective in autophagy, is more susceptible to TB infection and second by our previous demonstration that many autophagy genes are targets of IRF8. Consistent with this thinking, our immunostaining analysis revealed that NTM ingested within macrophages are captured by autophagosomes. In WT cells these processes can be visualized by LC3 puncta, a fluorescent autophagosome marker. However, Irf8 KO macrophages, where LC3 expression is low, failed to form these punctate structures and did not associate with NTM.
Click image to enlarge.
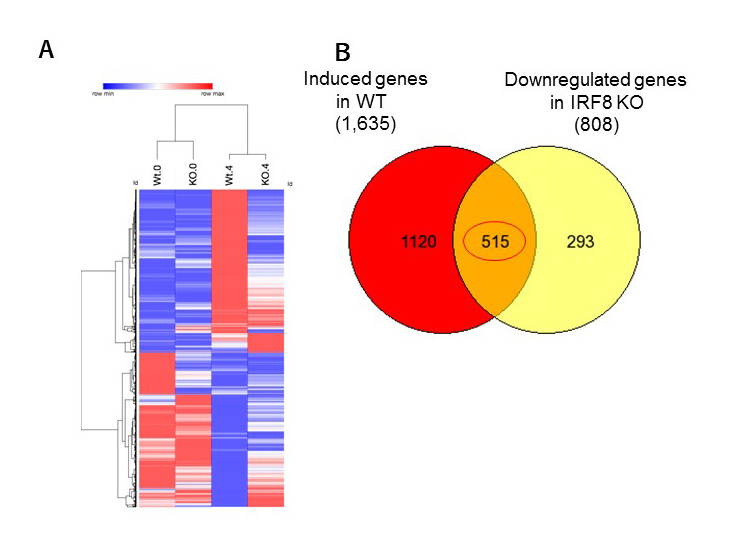
Figure 1. IRF8 KO macrophages are defective in innate response to mycobacterium infection.
RNA-seq analysis was performed for WT and IRF8 KO macrophages infected with NTM for 4 days.
Left: Hierarchical clustering of genes up- or down-regulated after NTM infection. The levels of RNA expression are shown by color gradients (References 1,2,3,4).
Right: Venn diagram showing the number of genes induced by NTM infection, but that are downregulated in IRF8 KO macrophages. KO macrophages failed to induce more than 30% of NTM response genes, including those well known for anti-microbial defense.
Hematopoietic stem cells depend on BRD4 for their maintenance and progenitor differentiation.
BRD4 is a bromodomain protein of the BET (bromodomain and extraterminal domain) family, which this laboratory has been studying for many years. BRD4 is expressed at high levels in most, if not all cells, and is necessary for very early embryonic development. Thus, conventionally created Brd4 knockout (KO) mice are embryonic lethal. BRD4 is a so-called “chromatin reader” owing to its binding to acetylated histones. It also recruits the transcription elongation factor P-TEFb, thus facilitating transcriptional elongation. Moreover, BRD4 has a critical role in forming super-enhancers. Super-enhancers are long stretches of regulatory DNAs densely occupied by transcription factors and chromatin regulators. They direct strong transcription of select genes and thus help define cellular and lineage identity. In the past several years, research on BRD4 has seen a dramatic upturn owing to the development of small-molecule inhibitors that inhibit binding of acetyl-histones to the BET family proteins. These inhibitors, affecting mostly the BET protein BRD4, antagonize cancer growth, particularly leukemia and lymphoma. Furthermore, BET inhibitors have been shown to inhibit inflammatory responses related to cardiovascular and autoimmune diseases. These reports implicate BRD4 in various disease processes and offer new therapeutic possibilities for several difficult-to-treat illnesses; indeed, clinical trials are being conducted for leukemia and inflammation. However, these developments present new issues stemming from the dearth of our understanding of the precise role of BRD4 in health and disease and of the mechanism of BRD4 action. Studies on inhibitors have inherent limitations due to uncertainty regarding their specificity, modality of action, and long-term consequences. For example, the impact of BET inhibitors on normal hematopoietic cells is not well understood, posing potential problems when treating blood cancers such as leukemia/lymphoma. BET inhibitor treatment may compromise the activity and maintenance of hematopoietic stem cells and may weaken the ability to combat infection, which is also relevant to treating inflammation, given that macrophages are the main effector of both inflammation and host defense.
We thus sought to gain a fuller understanding of BRD4’s activity in normal hematopoiesis and during inflammatory and innate immune responses. We studied Brd4 conditional knockout mice, focusing on hematopoiesis and macrophage responses. First, we tested mice in which Brd4 is deleted in early hematopoiesis by using the Vav-Cre technique. We showed that Brd4 KO mice die during fetal development owing to severe defects in the expansion of hematopoietic stem cells (HSC) and in the development of hematopoietic progenitor cells. As a consequence, Brd4 KO embryos fail to develop immune cells of all lineages, including lymphocytes and myeloid cells, which are important for innate and adaptive immunity. We also found that BRD4 is essential for the proliferation of macrophages, based on LysM-Cre–dependent deletion of Brd4 (LysM-Cre selectively targets macrophages and neutrophils); the resultant Brd4 KO mice failed to start IL-4–dependent peritoneal macrophage expansion. These results strongly point to a central role of BRD4 in immune cell expansion, required for maintaining immunity.
We investigated genome-wide distribution of BRD4 in macrophages in a resting condition and after LPS stimulation. LPS is a pathogen component that rapidly induces inflammatory genes and interferon-stimulated genes important for protection against pathogens. We found that BRD4 broadly occupies genic and intergenic regions. Within the genic region, BRD4 binding peaked at the transcription start site (TSS), although binding was detected over the 5′ promoter and within the coding regions. BRD4 binding over the genic regions markedly increased after LPS stimulation, indicating that BRD4 moves rapidly over the genome, presumably to accommodate a rapid alteration of histone acetylation. Furthermore, BRD4 displayed dense clustering over distant regulatory regions that represented super-enhancers. BRD4 clusters coincided with the H3K27 chromatin mark, which denotes super-enhancers as well as RNA polymerase II clustering. BRD4–containing super-enhancers localize to genes important for basic macrophage phenotypes and innate immune responses. Because it has been proposed that BRD4 is central for generation of super-enhancers, we were interested to know whether Brd4 KO macrophages possessed super-enhancers.
Click image to enlarge.
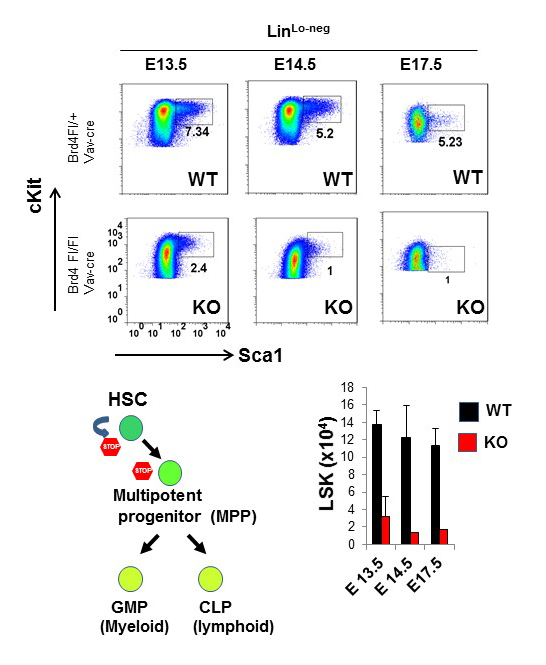
Figure 2. Hematopoietic stem cells depend on BRD4 for the self-renewal and progenitor differentiation.
Upper panel: Hematopoietic stem cells in fetal liver from WT and Brd4 KO embryos were identified at indicated days by flowcytometry.
Lower panel: (left) diagram showing the stages of HSC development. BRD4 is required for the earliest stage of hematopoiesis; (right) the total number of HSCs in WT and Brd4 KO embryos.
NSD2 guides interferon-induced H3.3 deposition and regulates transcription by recruiting the chromatin reader ZMYND.
Interferons (IFN) rapidly stimulate transcription of many IFN–stimulated genes (ISGs). We previously showed that ISG transcription is coupled with H3K36 trimethylation and deposition of the histone variant H3.3, creating stable epigenetic marks on ISGs. NSD2 (WHSC1, MMSET1) is a histone methyltransferase that methylates H3K36. We showed that global distribution and IFN–induced deposition of H3.3 was abrogated in mouse embryonic fibroblasts from Nsd2 KO mice, with only minor changes in the H3K36me3 distribution pattern. This led to aberrant ISG induction, in that a fraction of ISGs were constitutively expressed in Nsd2 KO cells even prior to IFN stimulation. Further, more than half the ISGs (about 200) were induced more rapidly and at higher levels in KO cells than in wild-type cells. ZMYND11 is a “chromatin reader” that was recently reported to recognize H3.3 and shown to repress transcription of some genes. We demonstrated that ZMYND11 is recruited to ISGs upon IFN stimulation and that recruitment is inhibited in Nsd2 KO cells. Consistent with these data, exaggerated ISG expression was observed in ZMYND11 KO cells. Our results reveal that ISG induction is internally coupled to NSD2-ZMYND11–dependent transcriptional suppression, which restrains transcriptional overdrive. ZMYND11–mediated repression may be linked to epigenetic memory for ISGs.
Click image to enlarge.
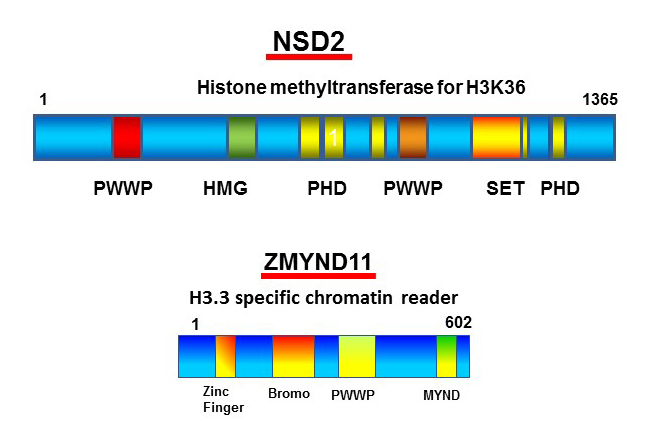
Figure 3. NSD2 guides interferon-induced H3.3 deposition and regulates transcription by recruiting the chromatin reader ZMYND.
Schematic diagram of NSD2 and ZMYND. NSD2 is a histone methyltranferase that is involved in dimethylation of H3K36 through the SET domain. We found that interferon-induced H3.3 deposition requires other domains of NSD2; thus H3.3 deposition is abrogated in Nsd2 KO cells. ZMYND was also recruited to transcribed genes in a NSD2– and H3.3–dependent manner. Our results provide mechanistic insight into the chromatin-marking event and its immediate impact on transcription.
Publications
- Wolf G, Rebollo R, Karimi MM, Ewing AD, Kamada R, Wu W, Wu B, Bachu M, Ozato K, Faulkner GJ, Mager DL, Lorincz MC, Macfarlan TS. On the role of H3.3 in retroviral silencing. Nature 2017 548:E1-E3.
- Mace EM, Bigley V, Gunesch JT, Chinn IK, Angelo LS, Care MA, Maisuria S, Keller MD, Togi S, Watkin LB, LaRosa DF, Jhangiani SN, Muzny DM, Stray-Pedersen A, Coban Akdemir Z, Smith JB, Hernandez-Sanabria M, Le DT, Hogg GD, Cao TN, Freud AG, Szymanski EP, Savic S, Collin M, Cant AJ, Gibbs RA, Holland SM, Caligiuri MA, Ozato K, Paust S, Doody GM, Lupski JR, Orange JS. Biallelic mutations in IRF8 impair human NK cell maturation and function. J Clin Invest 2017 127:306-320.
- Valanparambi, RM, Tam M, Gros PP, Auger JP, Segura M, Gros P, Jardim A, Geary TG, Ozato K, Stevenson MM. IRF-8 regulates expansion of myeloid-derived suppressor cells and Foxp3+ regulatory T cells and modulates Th2 immune responses to gastrointestinal nematode infection. PLoS Pathogens 2017 13:e1006647.
- Miyagawa F, Tagaya Y, Ozato K, Asada H. Essential requirement for IFN regulatory factor 7 in autoantibody production but not development of nephritis in murine lupus. J Immunol 2016 197:2167-2176.
- Bachu M, Dey A, Ozato K. Chromatin landscape of the IRF genes and role of the epigenetic reader BRD4. J Interferon Cytokine Res 2016 36:470-475.
Collaborators
- Steven Holland, MD, Laboratory of Clinical Infectious Diseases, NIAID, Bethesda, MD
- Katrin D. Mayer-Barber, Dr rer nat, Laboratory of Clinical Infectious Diseases, NIAID, Bethesda, MD
- Herbert Morse II, MD, Laboratory of Immunopathology, NIAID, Rockville, MD
- Dinah S. Singer, PhD, Experimental Immunology Branch, NCI, Bethesda, MD
- Tomohiko Tamura, MD, PhD, Tokyo University, Tokyo, Japan
- Jun Zhu, PhD, DNA Sequencing and Genomics Core, NHLBI, Bethesda, MD
Contact
For more information, email ozatok@mail.nih.gov or visit http://ozatolab.nichd.nih.gov.