Molecular Genetics of Heritable Human Disorders

- Janice Y. Chou, PhD, Head, Section on Cellular Differentiation
- Lisa Zhang, PhD, Staff Scientist
- Irina Arnaoutova, PhD, Staff Scientist
- Jun Ho Cho, PhD, Visiting Fellow
- Goo Young Kim, PhD, Visiting Fellow
- Joon Hyun Kwon, PhD, Visiting Fellow
- Javier Anduaga, BS, Technical Intramural Research Training Award Fellow
- Kurt Berckmueller, BS, Predoctoral Intramural Research Training Award Fellow
- Vamsi Reddy, BS, Predoctoral Intramural Research Training Award Fellow
- Brian C. Mansfield, PhD, Guest Researcher
We conduct research to delineate the pathophysiology and develop novel therapies for the three subtypes of type I glycogen storage disease (GSD-I): GSD-Ia, GSD-Ib, and GSD-Irs (GSD-I related syndrome). GSD-Ia is caused by a deficiency in glucose-6-phosphatase-α (G6Pase-α or G6PC), GSD-Ib is by a deficiency in the glucose-6-phosphate (G6P) transporter (G6PT or SLC37A4), and GSD-Irs, also known as severe congenital neutropenia syndrome type 4, by a deficiency in G6Pase-β. G6Pase-α and G6Pase-β are endoplasmic reticulum (ER)–bound G6P hydrolases, with active sites lying inside the lumen, which depend upon G6PT to translocate G6P from the cytoplasm into the ER lumen. The G6PT/G6Pase-α complex maintains interprandial glucose homeostasis while the G6PT/G6Pase-β complex maintains energy homeostasis and functionality of neutrophils. GSD-Ia and GSD-Ib patients manifest a common metabolic phenotype of impaired glucose homeostasis not shared by GSD-Irs. GSD-Ib and GSD-Irs patients manifest a common myeloid phenotype of neutropenia and myeloid dysfunction not shared by GSD-Ia. Inactivation of G6PT or G6Pase-β in neutrophils leads to enhanced apoptosis, which underlies neutropenia in GSD-Ib and GSD-Irs. A deficiency in either G6PT or G6Pase-β in neutrophils prevents recycling of glucose from the ER to the cytoplasm, leading to the impaired energy homeostasis that underlies neutrophil dysfunction in GSD-Ib and GSD-Irs. There is no cure for GSD-Ia, GSD-Ib, or GSD-Irs. Animal models of the three disorders are available and are being exploited to both delineate the disease more precisely and develop new treatment approaches, including gene therapy. We recently generated several efficacious G6Pase-α–expressing recombinant adeno-associated virus (rAAV) vectors and provided a proof-of-principle gene therapy in murine GSD-Ia that is safe, efficacious, and appropriate for entering clinical trials. Working with our commercial and clinical collaborators, we are expecting to initiate phase I/II clinical trials for human GSD-Ia in 2018/2019.
Molecular mechanism preventing hepatocellular adenoma and carcinoma (HCA/HCC) in GSD-Ia mice receiving gene therapy
The predominant subtypes of HCA in GSD-Ia are inflammatory HCA (IHCA, 52%) and β-catenin–mutated HCA (bHCA, 28%). We previously showed that non–tumor bearing (NT), rAAV–treated GSD-Ia mice (AAV-NT mice) expressing a wide range (0.9–63%) of normal hepatic G6Pase-α activity maintain glucose homeostasis and display physiologic features mimicking animals living under calorie restriction. We showed that in AAV-NT mice, the signaling pathways of the calorie restriction mediators AMPK and SIRT1 were activated, leading to inhibition of the activity of STAT3 and NFκB, pro-inflammatory and cancer-promoting transcription factors. SIRT1 also inhibits cancer metastasis by increasing the expression of E-cadherin, a tumor suppressor, and reducing the expression of mesenchymal markers. Consistently, in AAV-NT mice, hepatic levels of active STAT3 and of the p65 subunit of nuclear factor κB (NFκB) were reduced, as were expression of mesenchymal markers, STAT3 targets, NFκB targets, and β-catenin targets. AAV-NT mice also expressed elevated levels of E-cadherin and fibroblast growth factor 21 (FGF21), targets of SIRT1, and β-klotho, which can act as a tumor suppressor. Importantly, treating AAV-NT mice with a SIRT1 inhibitor markedly reversed many of the observed anti-inflammatory/anti-tumorigenic signaling pathways. In summary, activation of hepatic AMPK/SIRT1 and FGF21/β-klotho signaling pathways combined with down-regulation of STAT3/NFκB–mediated inflammatory and tumorigenic signaling pathways can explain the absence of hepatic tumors in AAV-NT mice.
Hepatic G6Pase-α activity is required to prevent HCA/HCC in GSD-Ia.
The hallmarks of GSD-Ia are impaired glucose homeostasis and long-term risk of hepatocellular adenoma and carcinoma (HCA/HCC). We previously developed a G6Pase-α–expressing rAAV vector, rAAV-G6PC, and showed that rAAV-G6PC–treated G6pc–/– mice expressing 3–63% of normal hepatic G6Pase-α activity (AAV mice) maintain glucose homeostasis and do not develop HCA/HCC. However, the threshold of hepatic G6Pase-α activity required to prevent tumor formation remained unknown. To increase the efficacy of the gene transfer vector, we constructed rAAV-co-G6PC, a rAAV vector expressing a codon-optimized (co) G6Pase-α and showed that rAAV-co-G6PC was more efficacious than rAAV-G6PC in directing hepatic G6Pase-α expression. Over an 88-week study, we showed that both rAAV-G6PC– and rAAV-co-G6PC–treated G6pc–/– mice expressing 3–33% of normal hepatic G6Pase-α activity maintained glucose homeostasis, lacked HCA/HCC, and were protected against age-related obesity and insulin resistance. Of the eleven rAAV-G6PC/rAAV-co-G6PC–treated G6pc–/– mice harboring 0.9–2.4% of normal hepatic G6Pase-α activity (AAV-low mice), three expressing 0.9–1.3% of normal hepatic G6Pase-α activity developed HCA/HCC, while eight did not (AAV-low-NT). We also showed that the AAV-low-NT mice exhibited a phenotype indistinguishable from that of AAV mice expressing 3% or more of normal hepatic G6Pase-α activity. The results establish the threshold of hepatic G6Pase-α activity required to prevent HCA/HCC and show that GSD-Ia mice harboring less than 2% of normal hepatic G6Pase-α activity are at risk for tumor development.
Downregulation of SIRT1 signaling underlies hepatic autophagy impairment in GSD-Ia.
The most severe long-term complication in GSD-Ia is HCA/HCC of unknown etiology. The global G6pc–/– mice die early, well before HCA/HCC can develop, making studies on the mechanism of HCA/HCC in these mice difficult. We therefore generated liver-specific G6pc knock-out (L-G6pc–/–) mice, which survive to adulthood and develop HCA. A recent report showed that G6Pase-α deficiency causes impairment in autophagy, a recycling process important for cellular metabolism. However, the underlying mechanism is unclear. We showed that liver-specific knockout of G6Pase-α led to downregulation of SIRT1 signaling, which activates autophagy via deacetylation of autophagy-related (ATG) proteins, and of the FoxO family of transcriptional factors, which trans-activate autophagy genes. Consistently, defective autophagy in G6Pase-α–deficient liver was characterized by attenuated expressions of autophagy components, increased acetylation of ATG5 and ATG7, decreased conjugation of ATG5 and ATG12, and reduced autophagic flux. We further showed that hepatic G6Pase-α deficiency resulted in activation of ChREBP, a lipogenic transcription factor, increased expression of PPAR-γ, a lipid regulator, and suppressed expression of PPAR-α, a master regulator of fatty acid β-oxidation, all contributing to hepatic steatosis and downregulation of SIRT1 expression. An adenovirus vector–mediated increase in hepatic SIRT1 expression corrected autophagy defects but failed to rectify metabolic abnormalities associated with G6Pase-α deficiency. Importantly, rAAV vector–mediated restoration of hepatic G6Pase-α expression corrected metabolic abnormalities, restored SIRT1-FoxO signaling, and normalized defective autophagy (see Figure). Taken together, the data show that hepatic G6Pase-α deficiency–mediated down-regulation of SIRT1 signaling underlies defective hepatic autophagy in GSD-Ia.
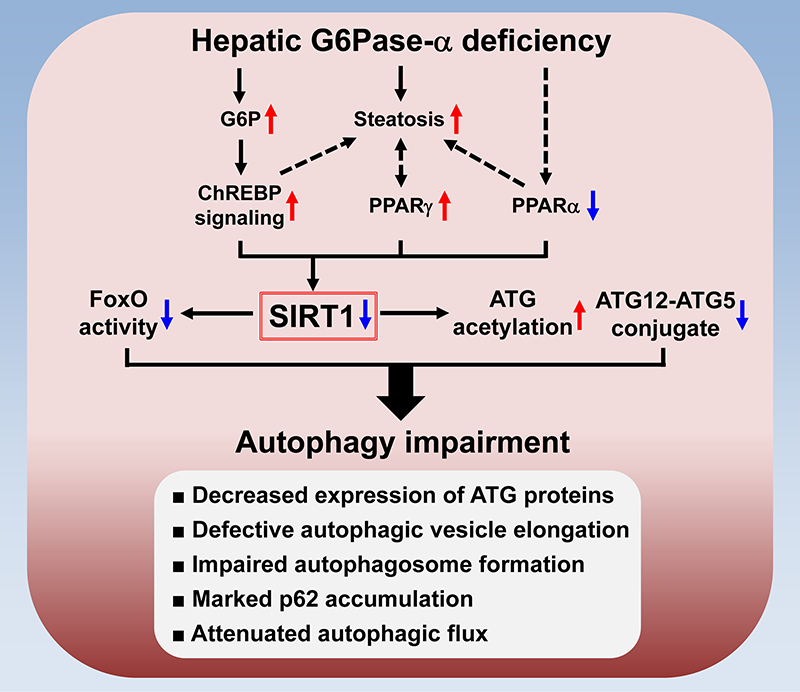
Click image to enlarge.
The mechanism underlying autophagy impairment in hepatic G6Pase-α deficiency
Hepatic G6Pase-α deficiency leads to metabolic alterations including G6P accumulation and suppressed expression of PPARα, a master regulator of fatty acid β-oxidation. The G6P–mediated activation of ChREBP signaling induces lipogenesis, leading to hepatic steatosis, which increases the expression of PPARγ, another lipogenic factor. Moreover, aberrant PPARγ overexpression aggravates hepatic steatosis. The net outcome is downregulation of hepatic SIRT1 signaling. Impaired SIRT1 signaling increases ATG acetylation and reduces ATG12–ATG5 conjugation along with downregulation of FoxO signaling, which induces autophagy genes. Accordingly, hepatic G6Pase-α deficiency–mediated autophagy impairment is characterized by reduced expression of ATG proteins, defective autophagic vesicle elongation, impaired autophagosome formation, marked accumulation of the p62 adaptor protein (implicated in selective autophagy), and attenuated autophagic flux.
Liver-directed gene therapy for murine GSD-Ib
The G6pt–/– mice manifest both metabolic and myeloid dysfunction characteristic of human GSD-Ib. When left untreated, the G6pt–/– mice rarely survive weaning, reflecting the juvenile lethality seen in human GSD-Ib patients. Studies have shown that the choice of transgene promoter can impact targeting efficiency, tissue-specific expression, and the level of immune response or tolerance to the therapy. We therefore examined the safety and efficacy of liver-directed gene therapy in G6pt–/– mice using rAAV-GPE-G6PT and rAAV-miGT-G6PT, two G6PT–expressing rAAV8 vectors directed by the human G6PC and G6PT promoter/enhancer, respectively. Both vectors corrected hepatic G6PT deficiency in murine GSD-Ib but the G6PC promoter/enhancer was more efficacious. Over a 78-week study, we showed that G6pt–/– mice expressing 3–62% of normal hepatic G6PT activity exhibited a normalized liver phenotype. Two of the 12 mice expressing less than 6% of normal hepatic G6PT activity developed HCA. All treated mice were leaner and more sensitive to insulin than wild-type mice. Mice expressing 3–22% of normal hepatic G6PT activity exhibited higher insulin sensitivity than mice expressing 44–62%. The levels of insulin sensitivity correlated with the magnitude of hepatic ChREBP signaling activation. In summary, we established the threshold of hepatic G6PT activity required to prevent tumor formation and showed that mice expressing 3–62% of normal hepatic G6PT activity maintained glucose homeostasis and were protected against age-related obesity and insulin resistance.
GSD-Ib neutrophils exhibit impaired cell adhesion and migration.
GSD-Ib, caused by a deficiency in G6PT, is characterized by impaired glucose homeostasis, myeloid dysfunction, and long-term risk of HCA. Neutrophils play an essential role in the defense against invading pathogens. The recruitment of neutrophils towards the inflammation sites in response to inflammatory stimuli is a tightly regulated process involving rolling, adhesion, and transmigration. We investigated the role of G6PT in neutrophil adhesion and migration using in vivo and in vitro models. We showed that G6pt–/– (GSD-Ib) mice manifested severe neutropenia in both blood and bone marrow, and that treating G6pt–/– mice with granulocyte colony–stimulating factor (G-CSF) corrected neutropenia. However, upon thioglycolate challenge, neutrophils from both untreated and G-CSF–treated G6pt–/– mice exhibited lowered ability to migrate to the peritoneal cavity. In vitro migration and cell adhesion of G6PT–deficient neutrophils were also significantly impaired. Defects in cell migration were not the result of enhanced apoptosis or altered fMLP receptor expression. Remarkably, the expression of the β2 integrins CD11a and CD11b, which are critical for cell adhesion, was greatly reduced in G6PT–deficient neutrophils. The study suggests that deficiencies in G6PT cause impairment in neutrophil migration and adhesion via aberrant expression of β2 integrins; our finding should facilitate the development of novel therapies for GSD-Ib.
Additional Funding
- The Children's Fund for Glycogen Storage Disease Research, 2016
- Dimension Therapeutics (Cambridge, MA) under a Cooperative Research and Development Agreement (CRADA)
Publications
- Kim GY, Lee YM, Kwon JH, Cho JH, Mansfield BC, Chou JY. Downregulation of pathways implicated in liver inflammation and tumorigenesis of glycogen storage disease type Ia mice receiving gene therapy. Hum Mol Genet 2017 26:1890-1899.
- Cho J-H, Kim GY, Pan CJ, Anduaga J, Choi E-J, Mansfield BC, Chou JY. Downregulation of SIRT1 signaling underlies hepatic autophagy impairment in glycogen storage disease type Ia. PLOS Genet 2017 13:e1006819.
- Kim GY, Lee YM, Kwon JH, Cho JH, Pan CJ, Starost MF, Mansfield BC, Chou JY. Glycogen storage disease type Ia mice with less than 2% of normal hepatic glucose-6-phosphatase-alpha activity restored are at risk of developing hepatic tumors. Mol Genet Metab 2017 120:229-234.
- Kim GY, Lee YM, Kwon JH, Jun HS, Chou J. Glycogen storage disease type Ib neutrophils exhibit impaired cell adhesion and migration. Biochem Biophys Res Commun 2017 482:569-574.
- Kwon JH, Lee YM, Cho J-H, Kim GY, Anduaga, J, Starost MF, Mansfield BC, Chou JY. Liver-directed gene therapy for murine glycogen storage disease type Ib. Hum Mol Genet 2017 26(22):4395-4405.
Collaborators
- Eui-Ju Choi, PhD, Korea University, Seoul, South Korea
- Alessandra Eva, PhD, Istituto Giannina Gaslini, Genova, Italy
- Hyun Sik Jun, PhD, Korea University, Seoul, South Korea
- Luigi Varesio, PhD, Istituto Giannina Gaslini, Genova, Italy
- David A. Weinstein, MD, MSc, UConn School of Medicine, Farmington, CT
Contact
For more information, email chou@helix.nih.gov or visit https://irp.nih.gov/pi/janice-chou.