Evolving Mechanisms of Transcriptional Control in Mammals

- Todd S. Macfarlan, PhD, Head, Unit on Mammalian Epigenome Reprogramming
- Carson Miller, PhD, Postdoctoral Fellow
- Gernot Wolf, PhD, Visiting Fellow
- Peng Yang, PhD, Visiting Fellow
- Sherry Ralls, BA, Biologist
- Don Hoang, BS, Postbaccalaureate Fellow
- Drew Honson, BS, Postbaccalaureate Fellow
- Angela Maggio, BS, Postbaccalaureate Fellow
- Matthew Tinkham, BS, Postbaccalaureate Fellow
- Justin Demmerle, BA, Graduate Student
- Alyssa Kosturakis, MSc, Medical Research Scholars Program Fellow
- Yixuan Wang, PhD, Collaborator
The “epigenome” refers to the heritable chemical changes in DNA and histone proteins that can be passed on through cell division and potentially across generations. Dramatic reprogramming of the epigenome begins at conception and continues throughout embryonic and postnatal development, underlying the gene-expression changes that drive the generation of diverse cell types that make up complex multicellular organisms. By exploring transcription factors that recognize parasitic mobile DNA elements to initiate stable heterochromatic silencing, we study how the epigenetic state of mammals is initially established in early development. We also study how one large family of these parasitic elements, the endogenous retroviruses (ERVs), play an important role in the development and evolution of new traits in mammals by rewiring gene expression networks. We also explore how the epigenetic state of a cell works in conjunction with combinatorial codes of transcription factors to generate the huge diversity of cell types required for mammalian development.
Retroviruses pose a threat to human health by infecting somatic cells, but retroviruses have also been infecting our mammalian ancestors for millions of years, accumulating in the germ-line as ERVs that account for about 10% of our genomic DNA. The laboratory studies ERVs from two perspectives: first as parasites that must be kept in check by the host to prevent widespread viral activation and second, as symbionts that can be co-opted by the host for evolutionary advantage. Our long-term objective is to understand how the host has adapted recognition machinery to establish epigenetic silencing of ERVs, how ERVs sometimes evade these silencing mechanisms, and how this “arms race” between ERVs and the host have led to co-option of viral regulatory sequences that may have contributed to the evolution of mammals. We hypothesize that the rapidly diversifying KRAB-ZFP family (see below) plays a critical role in the recognition and silencing of ERVs and nearby genes, and we are taking systematic genetic and biochemical approaches to explore this function.
Kruppel-associated box zinc finger proteins (KRAB-ZFPs) have emerged as candidates that recognize ERVs. KRAB-ZFPs are rapidly evolving transcriptional repressors that emerged in a common ancestor of coelacanth, birds, and tetrapods. They make up the largest family of transcription factors in mammals (estimated to be several hundred in mice and humans). Each species has its own unique repertoire of KRAB-ZFPs, with a small number shared with closely related species and a larger fraction specific to each species. Despite their abundance, little is known about their physiological functions. KRAB-ZFPs consist of an N-terminal KRAB domain that binds to the co-repressor KAP1 and a variable number of C-terminal C2H2 zinc finger domains that mediate sequence-specific DNA binding. KAP1 directly interacts with the KRAB domain, which recruits the histone methyltransferase (HMT) SETDB1 and heterochromatin protein 1 (HP1) to initiate heterochromatic silencing. Several lines of evidence point to a role for the KRAB-ZFP family in ERV silencing. First, the number of C2H2 zinc-finger genes in mammals correlates with the number of ERVs. Second, the KRAB-ZFP protein ZFP809 was isolated based on its ability to bind to the primer binding site for proline tRNA (PBSPro) of murine leukemia virus (MuLV). Third, deletion of the KRAB-ZFP co-repressors Trim28 or Setdb1 leads to activation of many ERVs. We have thus begun a systematic interrogation of KRAB-ZFP function as a potential adaptive repression system against ERVs.
Given that it was originally identified as part of a repression complex that recognizes infectious MuLV via direct binding to the 18 nt Primer Binding Site for Proline (PBSpro) sequence, we focused on ZFP809 as a likely ERV–suppressing KRAB-ZFP. We hypothesized that ZFP809 might function in vivo to repress other ERVs that utilized the PBSpro. Using ChIP-seq of epitope-tagged ZFP809 in embryonic stem cells (ESCs) and embryonic carcinoma (EC) cells, we determined that ZFP809 bound to several sub-classes of ERV elements via the PBSpro. We generated Zfp809 knockout mice to determine whether ZFP809 was required for VL30pro silencing. We found that Zfp809 knockout tissues displayed high levels of VL30pro elements and that the targeted elements display an epigenetic shift from repressive epigenetic marks (H3K9me3 and CpG methylation) to active marks (H3K9Ac and CpG hypo-methylation). ZFP809–mediated repression extended to a handful of genes that contained adjacent VL30pro integrations. Furthermore, using a combination of conditional alleles and rescue experiments, we determined that ZFP809 activity was required in development to initiate silencing, but not in somatic cells to maintain silencing. The studies thus provided the first demonstration for the in vivo requirement of a KRAB-ZFP in the recognition and silencing of ERVs.
As a follow-up to our studies on ZFP809, we have begun a systematic analysis of KRAB-ZFPs using a medium-throughput ChIP-seq screen and functional genomics of KRAB-ZFP clusters and individual KRAB-ZFP genes. Our ChIP-seq data demonstrate that the majority of recently evolved KRAB-ZFP genes interact with and repress distinct and partially overlapping ERV targets. This is supported by a recent knockout mouse line lacking about 17 KRAB-ZFPs (generated with CRISPR/Cas9 engineering) that displays an ERV reactivation phenotype.
Like ZFP809, KRAB-ZFPs initiate ERV silencing by establishing methylation of histone H3 lysine 9 via the recruitment of the histone methyltransferase SETDB1. Mice have three histone H3 variants (H3.1, H3.2, and H3.3), and we set out to explore whether one or more of these variants is critical for ERV silencing. Using ChIP-seq in primary mouse embryonic fibroblasts and induced pluripotent stem cells containing genetically tagged histone H3.3 genes, we found a strong enrichment of the variant histone H3.3 at ERVs co-occupied by KAP1, SETDB1, and H3K9me3, including VL30Pro elements recognized by ZFP809. Importantly, this enrichment was present only in pluripotent cells. We therefore explored the possibility that the deposition of histone H3.3 is required for ERV silencing. To test this hypothesis, we used CRISPR/Cas9 to create a homozygous “floxed” Daxx gene, given that DAXX had previously been shown to be responsible for H3.3 deposition at telomeres in ESCs. We found that acute loss of Daxx by Cre-mediated recombination in ESCs caused a complete loss of histone H3.3 at ERVs, but had little effect on ERV reactivation in contrast to deletion of Setdb1, which leads to massive ERV reactivation. These data suggest that DAXX–dependent deposition of histone H3.3 is dispensable for ERV silencing, a finding that is in conflict with a recent report arguing that some ERVs display reactivation in histone H3.3 KO ESCs, and that IAP elements, in particular, become retro-transpositionally active. Our re-analysis of this dataset challenges this central conclusion. We found that there is no correlation between ERVs marked by histone H3.3 and those showing reactivation in H3.3 KOs; furthermore, we demonstrated that the reported IAP “re-integrations” are not the result of retrotransposition, but were simply polymorphic IAP elements mixed into the genetic background of the ESCs used for the study. The authors of the study have acknowledged this error in their interpretation. Our data support a model in which histone H3.3 is deposited into ERVs and other KRAB-ZFP target genes in pluripotent cells but that this deposition is not required for ERV silencing, a model that has been supported further in the recent literature.
Although our data show that many KRAB-ZFPs repress ERVs, we also found that more ancient KRAB-ZFPs, which emerged in a human/mouse common ancestor, do not bind to and repress ERVs. One of these KRAB-ZFPs, ZFP568, plays an important role in silencing a key developmental gene that may have played a critical role in the onset of viviparity in mammals. Using ChIP-seq and biochemical assays, we determined that ZFP568 is a direct repressor of a placental-specific isoform of the Igf2 gene called Igf2-P0. Insulin-like growth factor 2 (Igf2) is the major fetal growth hormone in mammals. We demonstrated that loss of Zfp568, which causes gastrulation failure, or mutation of the ZFP568 binding site at the Igf2-P0 promoter causes inappropriate Igf2-P0 activation. We also showed that this lethality could be rescued by deletion of Igf2. These data highlight the exquisite selectivity by which members of the KRAB-ZFP family repress their targets and identify an additional layer of transcriptional control of a key growth factor regulating fetal and placental development.
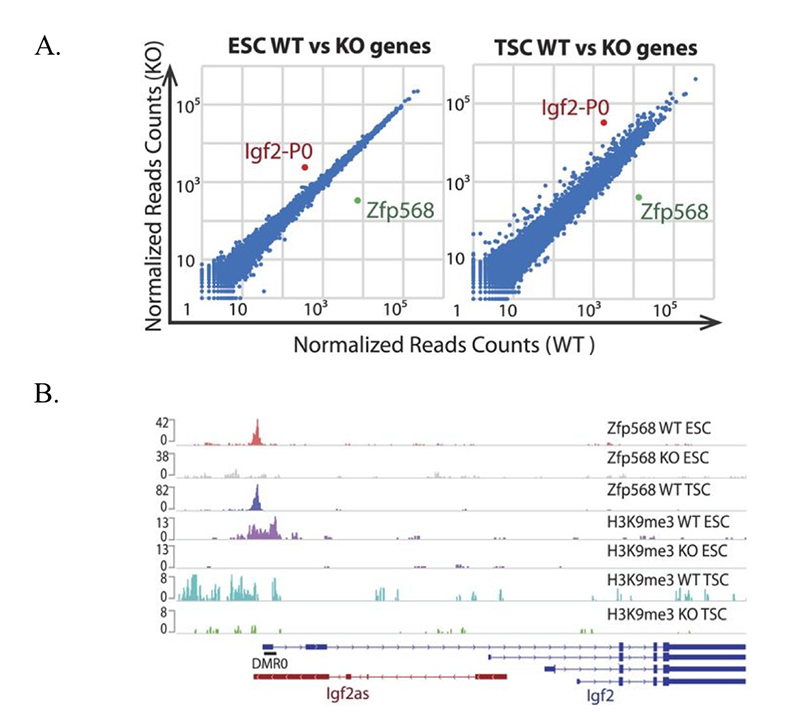
Click image to enlarge.
ZFP568 directly represses the Igf2-P0 transcript.
A. Scatter plots of gene expression in Zfp568 WT and KO ESCs and TSCs, as determined by RNA-seq.
B. ZFP568 and H3K9me3 ChIP-seq signals at the Igf2 locus in Zfp568 WT and KO ESCs and TSCs. DMR0 is a differentially methylated region overlapping exon 1 of the Igf2-P0 transcript. Igf2as is the Igf2 antisense transcript.
Additional Funding
- Human Placenta Project ($30K): “Quantifying placental gene expression from maternal plasma-isolated cell-free nucleic acids for real-time monitoring of placental health”
- NIH DDIR Award: “CRISPRi-scRNA-seq screening to systematically dissect retrotransposon silencing.” Role: Co-PI
Publications
- Wolf G, Rebollo R, Karimi MM, Ewing AD, Kamada R, Wu W, Wu B, Bachu M, Ozato K, Faulkner GJ, Mager DL, Lorincz MC, Macfarlan TS. On the role of H3.3 in retroviral silencing. Nature 2017 548E1-E3.
- Yang P, Wang Y, Hoang D, Tinkham M, Patel A, Sun M-A, Wolf G, Baker M, Chien H-C, Lai N, Cheng X, Shen C-K J, Macfarlan TS. A placental growth factor is silenced in mouse embryos by the zinc finger protein ZFP568. Science 2017 356 (6339):757-759.
- Yang P, Wang Y, Macfarlan TS. The role of KRAB-ZFPs in transposable element repression and mammalian evolution. Trends Genet 2017 33(1):871-881.
- Choi YJ, Lin CP, Risso D, Chen S, Kim TA, Tan MH, Li JB, Wu Y, Chen C, Xuan Z, Macfarlan T, Peng W, Lloyd K, Kim SY, Speed TP, He L. Deficiency of microRNA miR-34a expands cell fate potential in pluripotent stem cells. Science 2017 355(6325):eaag1927.
- Wang C, Lee J-E, Lai B, Macfarlan TS, Xu B, Zhuang L, Liu C, Peng W, Ge K. Enhancer priming by H4K4 methyltransferase MLL4 controls cell fate transition. Proc Natl Acad Sci USA 2016 113(42):11871-11876.
Collaborators
- Xiaodong Cheng, PhD, Emory University, Atlanta, GA
- Kai Ge, PhD, Adipocyte Biology and Gene Regulation Section, NIDDK, Bethesda, MD
- Lin He, PhD, University of California, Berkeley, CA
- Keiko Ozato, PhD, Section on Molecular Genetics of Immunity, NICHD, Bethesda, MD
- Che-Kun James Shen, PhD, Academia Sinica, Taipei, Taiwan
Contact
For more information, email todd.macfarlan@nih.gov or visit http://macfarlan.nichd.nih.gov.