Chromatin Remodeling and Gene Activation

- David J. Clark, PhD, Head, Section on Chromatin and Gene Expression
- Peter Eriksson, PhD, Staff Scientist
- Razvan V. Chereji, PhD, Research Fellow
- Josefina Ocampo, PhD, Visiting Fellow
- Sean C. Clark, MS, Postbaccalaureate Fellow
- Allison F. Dennis, BS, Graduate Student
Our basic goal is to understand how chromatin structure influences gene regulation. Chromatin is generally repressive in nature but its structure is manipulated by cells in a regulated way to determine which genes are potentially transcriptionally active and which genes remain repressed in a given cell type. The regulation depends on interactions between DNA sequence–specific transcription factors, chromatin enzymes, and chromatin.
The structural subunit of chromatin is the nucleosome core, which contains about 147 bp of DNA wrapped approximately 1.7 times around a central histone octamer composed of two molecules each of the four core histones (H2A, H2B, H3, and H4). Generally, nucleosomes are regularly spaced along the DNA, like beads on a string. At physiological salt concentrations, the beads-on-a-string structure folds spontaneously to form a fiber of about 30 nm width, assisted by the linker histone (H1), which binds to the nucleosome core and to the linker DNA. Thus, collectively, the histones control DNA accessibility.
Gene activation involves the recruitment of a set of factors to a promoter in response to appropriate signals, ultimately resulting in the formation of an initiation complex by RNA polymerase II (Pol II) and transcription. These events occur in the presence of nucleosomes, which are compact structures capable of blocking transcription at every step. To circumvent and regulate the chromatin block, eukaryotic cells possess dedicated enzymes, including ATP–dependent chromatin-remodeling machines, histone-modifying complexes and histone chaperones. The remodeling machines use ATP to move nucleosomes along or off DNA (e.g., the SWI/SNF, RSC, CHD, and ISWI complexes), or to exchange histone variants between nucleosomes (e.g., the SWR complex). The histone-modifying complexes contain enzymes that modify the histones post-translationally to alter their DNA–binding properties and to mark them for recognition by other complexes, which have activating or repressive roles (the "histone code" hypothesis). Histone-modifying enzymes include histone acetylases (HATs), deacetylases (HDACs), methylases, and kinases. Histone chaperones mediate histone transfer reactions that occur during transcription and DNA replication (e.g., Asf1 and the CAF-1 complex). These enzymes, together with DNA–methylating and de-methylating enzymes, are central to epigenetics.
Many human diseases have been linked to chromatin-remodeling enzymes and epigenetic modifications. For example, mutations in the hSNF5 subunit of the SWI/SNF complex are strongly linked to pediatric rhabdoid tumors. The CHD class of ATP–dependent remodelers has also been linked to cancer and to autism. Cancer therapies and drugs aimed at epigenetic targets are being tested. Recent studies have revealed a correlation between a linker histone variant and tumor heterogeneity. A full understanding of the functions of chromatin structure, enzymes, and modifications is therefore vital.
Non-histone barrier complexes occupy nucleosome-depleted promoters.
Our aim is to dissect chromatin-remodeling mechanisms in vivo and elucidate their contributions to gene regulation. Our current efforts are focused on elucidating the contributions of the various ATP–dependent chromatin-remodeling complexes to chromatin organization in vivo. Most of our work involves the use of budding yeast Saccharomyces cerevisiae as a model organism, but we are also involved in some mouse chromatin studies. During the past year, we made significant progress towards understanding the nature of promoter chromatin, more specifically, on the question of what occupies the nucleosome-depleted region that is characteristic of most yeast promoters and many mouse and human promoters. Our study (Reference 1) is summarized below.
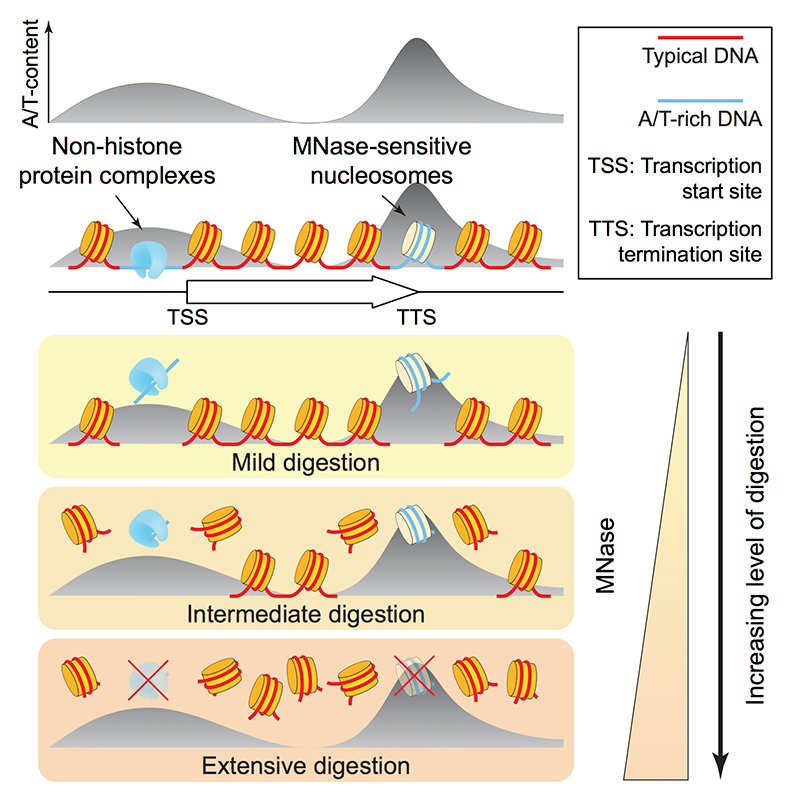
Most genes in yeast and in higher organisms have a characteristic chromatin organization, in which the promoter, just upstream of the transcription start site (TSS), is depleted of nucleosomes (the nucleosome-depleted region or NDR). The NDR is flanked by well positioned nucleosomes in phased arrays with a characteristic spacing. These observations are based primarily on experiments using micrococcal nuclease (MNase), which digests linker DNA rapidly but nucleosome core DNA much more slowly. A central question in our work is the nature of the NDR, whether it is protein-free or occupied by a large non-histone complex. An important factor is the DNA sequence itself: poly(dA) sequences are commonly found in promoters (though they are not universal) and exclude nucleosomes to some extent in vitro, an effect that is, however, relatively weak. Recently, several labs reported that many promoter NDRs are not actually nucleosome-free but occupied by easily digested, unstable “fragile nucleosomes.” However, other labs reported high-resolution mapping by ChIP and tiling microarray that provide little evidence for histones at yeast promoters. Micrococcal nuclease (MNase)–sensitive nucleosomes have also been reported in higher eukaryotes.
We addressed the important issue of the nature of the NDR in yeast (Reference 1). We confirmed that an MNase–sensitive complex is present at yeast promoters. The critical question is whether histones are present at NDRs, as predicted if fragile nucleosomes are formed at NDRs. We used two different approaches to detect histones H4 and H2B: MNase-ChIP-seq, which involves immuno-precipitation (IP) of nucleosomes from MNase digests; and standard ChIP-seq using sonication, which does not depend on the use of MNase. Both sets of data show that NDRs are strongly depleted of histones. Although we found no evidence of fragile nucleosomes at promoters, we did detect MNase–sensitive nucleosomes elsewhere in the genome. However, they have high A/T content, suggesting that MNase sensitivity does not indicate structural instability but a preference of MNase for A/T–rich DNA—simply, that A/T–rich nucleosomes are digested faster than G/C–rich nucleosomes. We confirmed our conclusions by analyzing ChIP-exo, chemical mapping, and ATAC-seq data from other labs. By analogy with stable RNA polymerase III transcription complexes at tRNA genes, which are composed of TFIIIB and TFIIIC, we propose that RNA polymerase II promoters are occupied by similar stable transcription complexes (Figure 1). Currently, we are endeavoring to identify the MNase–sensitive complexes that occupy promoter NDRs.
Click image to enlarge.
Figure 1. MNase–sensitive non-histone barrier complexes at promoters and MNase–sensitive nucleosomes containing AT–rich DNA at 3′-untranslated regions
Products of MNase digestion of chromatin: non-histone complexes (shown in blue) are located at promoters (at nucleosome-depleted regions, “NDRs”) and act as barriers to nucleosome formation (Reference 1).
MNase: micrococcal nuclease. TSS: transcription start site. TTS: transcription termination site (i.e., the poly-A addition site). Yellow discs are canonical nucleosomes.
Chromatin organization in trypanosomes
The compaction of DNA in chromatin in eukaryotes allowed the expansion of genome size and coincided with significant evolutionary diversification. However, chromatin generally represses DNA function, and mechanisms co-evolved to regulate chromatin structure and its impact on DNA. This included the selection of specific nucleosome positions to modulate accessibility to the DNA. Trypanosoma brucei, a member of the Excavates supergroup, falls into an ancient evolutionary branch of eukaryotes and provides valuable insight into the organization of chromatin in early genomes.
In a collaboration with the Patterton and Rundenko labs, we mapped nucleosome positions in both the bloodstream and the procyclic forms of T. brucei, using MNase-seq (Reference 2). We found that chromatin organization in T. brucei is quite different from that of other eukaryotes, namely the RNA polymerase II initiation regions in T. brucei do not exhibit pronounced nucleosome depletion and show little evidence for defined –1 and +1 nucleosomes. In contrast, a relatively well positioned nucleosome is formed on the splice acceptor sites within the polycistronic transcription units. The RNA polyadenylation sites are depleted of nucleosomes, with a single well positioned nucleosome present immediately downstream of the predicted sites. The regions flanking the silent variant surface glycoprotein (VSG) gene cassettes show extensive nucleosomal arrays, which may repress cryptic transcription initiation. The silent VSG genes themselves exhibit a less regular nucleosomal pattern in both bloodstream and procyclic-form trypanosomes. DNA replication origins, when present within silent VSG gene cassettes, have a distinct nucleosomal organization when compared with replication origins in other chromosomal core regions.
The proto-chromatosome
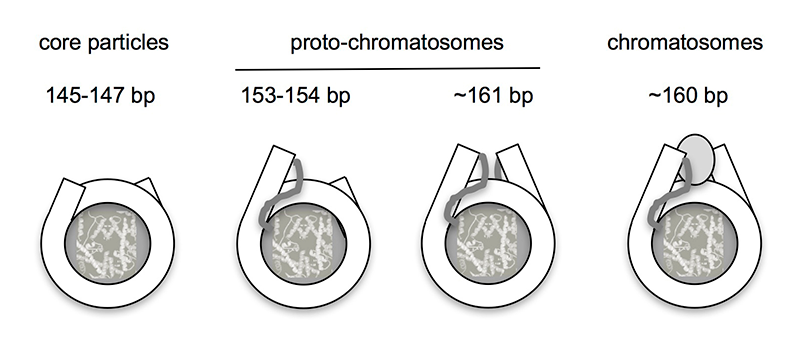
As described above, eukaryotic DNA is packaged into regularly spaced nucleosomes, resembling beads on a string, with each bead containing about 147 bp wrapped around a core histone octamer. Linker histone (H1) binds to the linker DNA to drive chromatin folding. Micrococcal nuclease (MNase) digestion studies reveal two mono-nucleosomal intermediates: the core particle (about 147 bp) and the chromatosome (about 160 bp; a core particle with additional DNA protected by H1). We recently developed an improved method for mapping nucleosomes, using exonuclease III to remove residual linker (MNase-Exo-seq). We discovered two new intermediate particles corresponding to core particles with about 7 bp of linker protruding from one side (about 154 bp) or both sides (about 161 bp), which are formed in the absence of H1. In a review article (Reference 3), we proposed that these “proto-chromatosomes” are stabilized by core histone–DNA contacts in the linker, about 7 bp from the nucleosome boundaries (Figure 2). The contacts may determine the topography of the H1–binding site, facilitating the binding of linker histone and chromatin folding.
Click image to enlarge.
Figure 2. Structural relationship between nucleosome core particles, proto-chromatosomes, and chromatosomes
Views from above, drawn approximately to scale, based on the nucleosome structure. The final 10 bp on each side of the nucleosome core are almost straight, projecting a short distance out of the particle. Proto-chromatosomes are shown with an extra 7 bp on one side (154 bp) or both sides (161 bp) with a continuing straight trajectory. We propose that the fundamental particle is the 161-bp proto-chromatosome, which has both extensions. We suggest that each extension is protected by a specific but unidentified core histone–DNA contact, most likely a histone tail (as indicated), to form the H1–binding site. The chromatosome is formed when H1 binds (shown here as a light gray oval in a symmetrical location, although this is controversial). From Reference 3.
Publications
- Chereji RV, Ocampo J, Clark DJ. MNase-sensitive complexes in yeast: nucleosomes and non-histone barriers. Mol Cell 2017 65:565-577.
- Maree JP, Povelones ML, Clark DJ, Rudenko G, Patterton HG. Well-positioned nucleosomes punctuate polycistronic pol II transcription units and flank silent VSG gene arrays in Trypanosoma brucei. Epigenetics Chromatin 2017 10:14.
- Ocampo J, Cui F, Zhurkin VB, Clark DJ. The proto-chromatosome—a fundamental subunit of chromatin? Nucleus 2016 7:382-387.
Collaborators
- Feng Cui, PhD, Rochester Institute of Technology, Rochester, NY
- Douglas Fields, PhD, Section on Nervous System Development and Plasticity, NICHD, Bethesda, MD
- Gordon L. Hager, PhD, Laboratory of Receptor Biology and Gene Expression, Center for Cancer Research, NCI, Bethesda, MD
- Alan G. Hinnebusch, PhD, Section on Nutrient Control of Gene Expression, NICHD, Bethesda, MD
- Philip R. Lee, PhD, Section on Nervous System Development and Plasticity, NICHD, Bethesda, MD
- Hugh G. Patterton, PhD, Stellenbosch University, Stellenbosch, South Africa
- Gloria Rundenko, PhD, Imperial College, London, United Kingdom
- Vasily M. Studitsky, PhD, Fox Chase Cancer Center, Temple University Health System, Philadelphia, PA
- Victor Zhurkin, PhD, Laboratory of Cell Biology, Center for Cancer Research, NCI, Bethesda, MD
Contact
For more information, email clarkda@mail.nih.gov or visit http://clarklab.nichd.nih.gov.