Regulation of Childhood Growth

- Jeffrey Baron, MD, Head, Section on Growth and Development
- Kevin Barnes, PhD, Senior Research Assistant
- Julian Lui, PhD, Staff Scientist
- Youn Hee Jee, MD, Clinical Scholar
- Audrey Lee, BS, Postbaccalaureate Fellow
- Shanna Yue, BA, Postbaccalaureate Fellow
Children grow taller because their bones grow longer. Bone elongation occurs at the growth plate, a thin layer of cartilage found near the ends of juvenile bones. In growth plates, new cartilage is produced through chondrocyte proliferation, hypertrophy, and cartilage matrix synthesis, and then newly formed cartilage is remodeled into bone. The process, termed endochondral ossification, results in bone elongation, which causes children to grow in height (linear growth). Consequently, mutations in genes that regulate growth-plate chondrogenesis cause abnormal bone growth and short stature in children. Depending on the severity and nature of the genetic abnormality, the phenotype can range from chondrodysplasias with short, malformed bones, to severe, often disproportionate, short stature, to mild proportionate short stature. If the genetic defect affects tissues other than the growth plate cartilage, the child may present with a more complex syndrome that includes other clinical abnormalities.
Many gene defects that affect the growth plate and thereby cause childhood growth disorders have been identified. However, for many children who are brought to medical attention for linear growth disorders, clinical, laboratory, and genetic evaluation fail to identify the underlying etiology. Genome-wide association studies and molecular studies of growth plate biology suggest that there are hundreds of genes that control linear growth. Therefore, there are likely many genetic causes of linear growth disorders that remain to be discovered.
We investigate the cellular and molecular mechanisms governing childhood growth and development. We focus particularly on growth at the growth plate. One goal of this work is to gain insight into the many human genetic disorders that cause childhood growth failure or overgrowth. A second goal is to develop new treatments for children with severe growth disorders.
Novel genetic causes of childhood growth disorders
To discover new genetic causes of childhood growth disorders, we invite families with monogenic growth disorders to the NIH Clinical Center, where we evaluate the clinical, biochemical, and radiological features of the condition. We then obtain DNA samples from informative family members and use powerful genetic approaches, including SNP (single-nucleotide polymorphism) arrays, to detect deletions, duplications, mosaicism, and uniparental disomy, combined with exome sequencing to detect single-nucleotide variants and small insertions/deletions in coding regions and splice sites. When sequence variants that are likely to cause the disorder are identified, we study in the laboratory the variants and the genes in which they occur to confirm that the variant is pathogenic, to elucidate the pathogenesis of the disorder, and to explore the role of the gene in normal growth.
These analysis have previously led to our studies of heterozygous mutations in ACAN, causing autosomal dominant short stature with advanced bone age and premature osteoarthritis [Reference 3; also Nilsson O et al., J Clin Endocrinol Metab 2014;99:E1510; and Gkourogianni A et al., 2017 J Clin Endocrinol Metab 2017;102:460] and to our study of biallelic mutations in BRF1 in children with growth failure, central nervous system abnormalities, and facial anomalies [Reference 4].
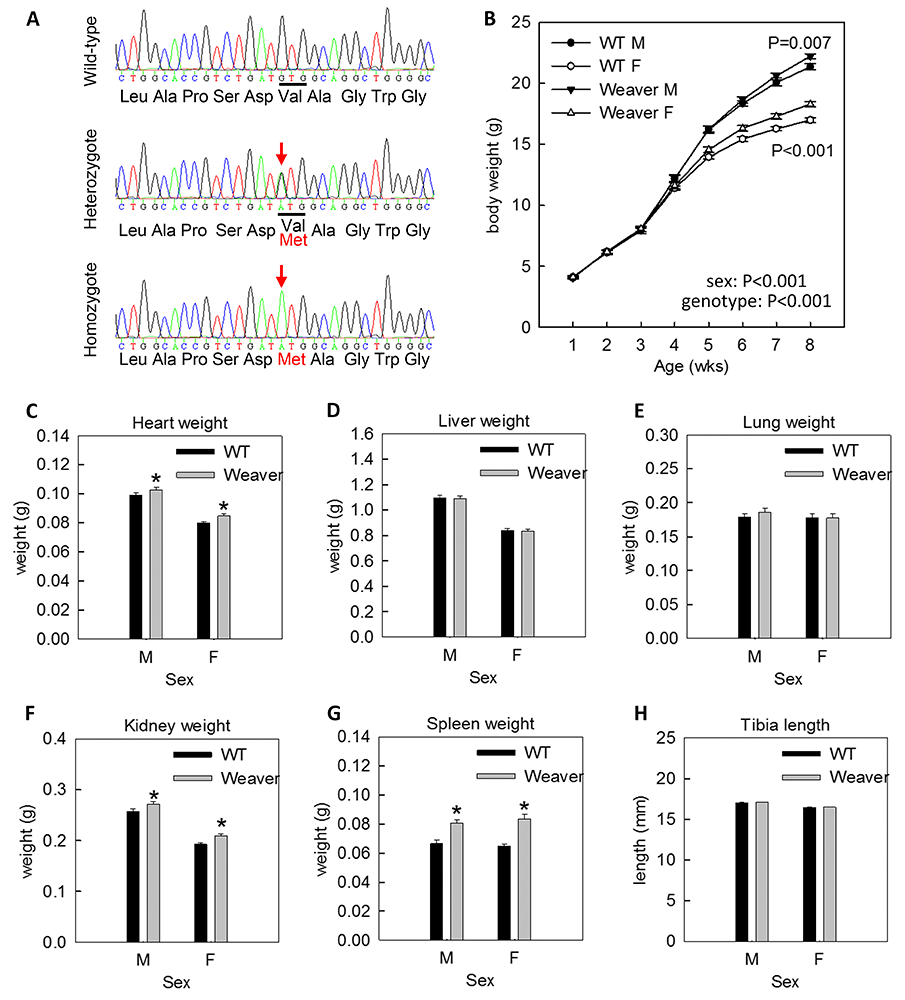
Click image to view.
Figure 1. Heterozygous Weaver mice showed postnatal overgrowth.
CRISPR/Cas9 genome editing was used to generate a mouse model carrying a Weaver-associated Ezh2 variant (V626M).
A. Crosses of heterozygotes generated mice that were wild-type, heterozygous, or homozygous for the Weaver variant, which was confirmed by PCR amplification of a genomic region containing the variant followed by Sanger sequencing.
B. Body weight was measured in heterozygous Weaver mice and wild-type littermates weekly up until 8 weeks of age (homozygotes were perinatally lethal). Two-way ANOVA showed that body weights differed significantly between sexes and genotypes. Pairwise comparison showed that, in both males and females, body weights were significantly increased in heterozygous Weaver mice.
C-H. At 8 weeks of age, heterozygote Weaver mice showed increased weights of heart, kidney, and spleen, but no differences in weights of liver and lung, or in tibia length, compared with wild-type mice.
We also studied a subject with tall stature, advanced bone age, and mild dysmorphic features [Reference 1]. Exome sequencing revealed a de novo missense mutation in EZH2. EZH2 encodes a histone methyltransferase that methylates histone 3 at lysine 27 (H3K27). The finding was consistent with previous reports that heterozygous missense mutations in EZH2 cause Weaver syndrome, which is characterized by tall stature, advanced bone age, characteristic facies, and variable intellectual disability. The molecular pathogenesis of the disorder is poorly understood. We previously showed that EZH2 plays a critical role in the regulation of chondrocyte proliferation and hypertrophy in the growth plate, which are the central determinants of skeletal growth [Lui JC, et al., Nat Commun 2016;7:13685]. To determine whether the EZH2 mutations found in Weaver syndrome cause a gain of function or a loss of function, EZH2 with the mutation found in our patient was expressed in mouse growth plate chondrocytes [Reference 1]. The mutant protein showed lower histone methyltransferase activity than the wild-type protein. The EZH2 mutation found in our subject was then introduced into mice using CRISPR/Cas9. Heterozygotes showed mild overgrowth, recapitulating the Weaver overgrowth phenotype (Figure 1). Both homozygous and heterozygous embryos showed decreased H3K27 methylation (Figure 2). The findings demonstrate that EZH2 mutations found in Weaver syndrome cause a partial loss of function.
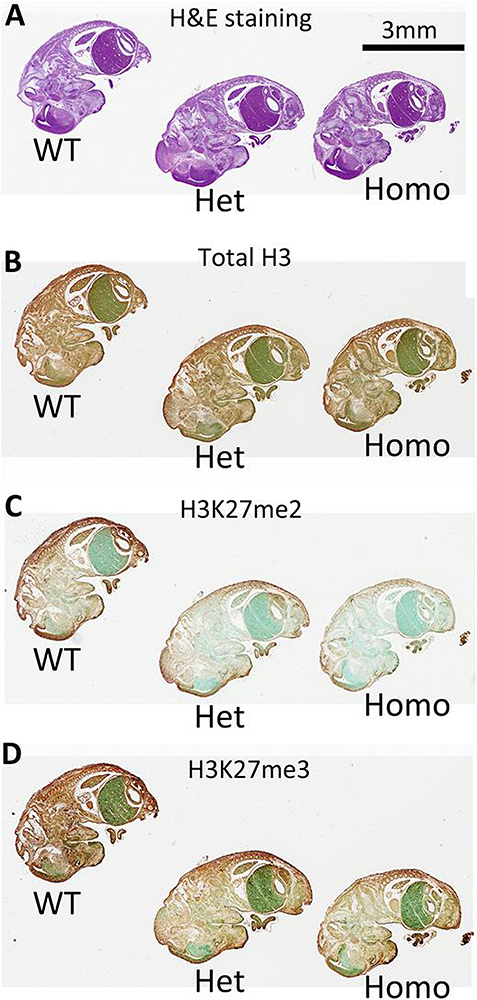
Click image to view.
Figure 2. Homozygous and heterozygous Weaver mice showed decreased H3K27 methylation in vivo.
Immunostaining of E14.5 embryos was used to examine difference in histone methyltransferase activity in vivo. Formalin-fixed wild-type, heterozygotes, and homozygotes were placed on the same slide and stained with hematoxylin and eosin for histology (A), antibody against total H3 (B), antibody against H3K27me2 (C), or antibody against H3K27me3 (D). The brown color indicates immunostaining, green represents counterstain. We found that both H3K27me2 and me3 levels were greatest in wild-type embryos, reduced in heterozygous embryos, and further reduced in homozygous embryos. No difference was observed for total H3 between different genotypes. Scale bar: 3mm.
Molecular and cellular mechanisms by which specific genes and pathways regulate childhood growth
For normal bone growth to occur, cells in the growth plate must differentiate from proliferative zone (PZ) chondrocytes to hypertrophic zone (HZ) chondrocytes. The important role of microRNAs (miRNAs) in growth plate chondrocyte differentiation was previously revealed by cartilage-specific ablation of Dicer, an enzyme essential for the biogenesis of many miRNAs. We sought to identify specific miRNAs that regulate differentiation of PZ chondrocytes to HZ chondrocytes [Reference 2]. First, we microdissected individual growth plate zones and performed miRNA profiling, using a solution hybridization method, as well as miRNA-seq (miRNA sequencing) to identify miRNAs that are preferentially expressed in PZ compared with HZ. We found that some of these preferentially expressed miRNAs (mir-374-5p, mir-379-5p, and mir-503-5p) serve to promote proliferation and inhibit hypertrophic differentiation. We also found evidence that the observed differential expression of mir-374-5p, mir-379-5p, and mir-503-5p between PZ and HZ is induced by the normal concentration gradient across the growth plate of parathyroid hormone–related protein (PTHrP). Taken together, our findings suggest that the PTHrP concentration gradient across the growth plate induces differential expression of mir-374-5p, mir-379-5p, and mir-503-5p between PZ and HZ. In PZ, the higher expression levels of these miRNAs promote proliferation and inhibit hypertrophic differentiation. In HZ, downregulation of these miRNAs inhibits proliferation and promotes hypertrophic differentiation.
Additional Funding
- NICHD Division of Intramural Research Director’s Award Project (2016-2018): The role of DLG2/PSD-93 in the initiation of puberty and the impact of mutations on NMDA receptor signaling and pubertal disorders
- NIH U01 award: 1U01HD086838-01A1 (2017-2021, ongoing): “Genetic Diagnosis of Childhood Growth Disorders”
Publications
- Lui JC, Barnes KM, Dong L, Yue S, Graber E, Rapaport R, Dauber A, Nilsson O, Baron J. Ezh2 mutations found in the Weaver overgrowth syndrome cause a partial loss of H3K27 histone methyltransferase activity. J Clin Endocrinol Metab 2018;103:1470-1478.
- Jee YH, Wang J, Jennings M, Clokie SJH, Nilsson O, Lui JC, Baron J. Mir-374-5p, mir-379-5p, and mir-503-5p regulate proliferation and hypertrophic differentiation of growth plate chondrocytes. Endocrinology 2018;59(3):1469-1478.
- Tatsi C, Gkourogianni A, Mohnike K, Dearment D, Witchel S, Andrade AC, Markello TC, Baron J, Nilsson O, Jee YH. Aggrecan mutations in non-familial short stature and short stature without accelerated skeletal maturation. J Endocr Soc 2017;1:1006-1011.
- Jee YH, Sowada N, Markello TC, Rezvani I, Borck G, Baron J. BRF1 mutations in a family with growth failure, markedly delayed bone age, and central nervous system anomalies. Clin Genet 2017;91(5):739-747.
- Giamanco NM, Jee YH, Wellstein A, Shriver CD, Summers TA, Baron J. Midkine and pleiotrophin concentrations in needle biopsies of breast and lung masses. Cancer Biomark 2017;20:299-307.
Collaborators
- Greti Aguilera, MD, Section on Endocrine Physiology, NICHD, Bethesda, MD
- Andrew Dauber, MD, Cincinnati Children’s Hospital, Cincinnati, OH
- Angela Delaney Freedman, MD, Office of the Clinical Director, NICHD, Bethesda, MD
- Dimiter Dimitrov, PhD, Laboratory of Experimental Immunology, Center for Cancer Research, NCI, Frederick, MD
- Thomas Markello, MD, PhD, Undiagnosed Diseases Program, NHGRI, Bethesda, MD
- Ola Nilsson, MD, PhD, Karolinska Institute, Stockholm, Sweden
- Katherine W. Roche, PhD, Receptor Biology Section, NINDS, Bethesda, MD
- Jack Yanovski, MD, PhD, Section on Growth and Obesity, NICHD, Bethesda, MD
Contact
For more information, email jeffrey.baron@nih.gov or visit baron.nichd.nih.gov/.