Childhood Neurodegenerative Lysosomal Storage Disorders

- Anil B. Mukherjee, MD, PhD, Head, Section on Developmental Genetics
- Maria B. Bagh, PhD, Research Fellow
- Abhilash Appu, PhD, Visiting Fellow
- Avisek Mondal, PhD, Visiting Fellow
- Tamal Sadhukhan, PhD, Visiting Fellow
- Sondra W. Levin, MD, Adjunct Scientist
- Zhongjian (Gary) Zhang, MD, PhD, Adjunct Scientist
- Sydney Casey, AB, Postbaccalaureate Intramural Research Training Award Fellow
The Section on Developmental Genetics conducts both basic laboratory research and clinical investigations into a group of the most common childhood neurodegenerative lysosomal storage disorders (LSDs), called neuronal ceroid lipofuscinoses (NCLs), also commonly known as Batten disease. The diseases affect mostly children and there is no effective treatment for any of the NCLs. Mutations in at least 13 different genes (called the CLNs) underlie various types of NCLs. Among these genes, CLN1, CLN2, CLN5, CLN10, and CLN13 encode soluble lysosomal enzymes; CLN4 and CLN14 encode peripherally associated cytoplasmic proteins; CLN11 encodes progranulin, a protein in the secretory pathway; and several transmembrane proteins with varying subcellular localizations are encoded by CLN3, CLN6, CLN7, CLN8, and CLN12.

Click image to view.
Section on Developmental Genetics Staff
Upper Left to Right: Sydney Casey, Abhilash Appu, Tamal Sadhukhan, and Maria Bagh.
Lower Left to Right: Sondra Levin, Gary Zhang, and Anil Mukherjee
Despite intense studies, the normal physiological functions of each of the CLN genes are poorly understood. Consequently, the development of mechanism-based therapeutic strategies remains challenging. Studies within the past decade have drastically changed our notion that the lysosomes are merely a terminal degradative organelle. Some of the emerging new roles of the lysosome include its central role in nutrient-dependent signal transduction regulating metabolism, cellular proliferation, or quiescence. Thus, endolysosomal dysfunction contributes to pathogenesis of virtually all LSDs.
Currently, our research focuses on understanding the molecular mechanisms of pathogenesis underlying infantile NCL (INCL: CLN1 disease), juvenile NCL (JNCL: CLN3 disease), and congenital NCL (CNCL: CLN10 disease). Interestingly, all 13 NCL types share some common clinical and pathologic features, such as intracellular accumulation of autofluorescent material, epileptic seizures, progressive psychomotor decline resulting predominantly from loss of cortical neurons in the cerebrum, neuro-inflammatory findings, visual impairment resulting from retinal degeneration, and shortened lifespan.
We first started investigating the INCL (CLN1 disease), which is caused by mutations in the CLN1 gene encoding a lysosomal depalmitoylating enzyme, palmitoyl-protein thioesterase-1 (PPT1). Numerous proteins in the body, especially in the brain, undergo post-translational modification called S-palmitoylation (also called S-acylation). In this process, a long-chain fatty acid is attached to specific cysteine residues in polypeptides via thioester linkage. While S-palmitoylation plays important roles in membrane anchorage of soluble proteins, protein-protein interaction, and protein stability, the proteins must also be depalmitoylated for recycling or degradation in lysosome. PPT1 catalyzes the cleavage of thioester linkage S-palmitoylated proteins, important because S-palmitoylated proteins are refractory to degradation by lysosomal hydrolases, and PPT1 deficiency leads to lysosomal accumulation of the lipidated proteins (constituents of ceroid), leading to the pathogenesis of INCL. Children afflicted with INCL are normal at birth but, by 11 to 18 months of age, they exhibit signs of psychomotor retardation. By 2 years of age, they are completely blind owing to retinal degeneration and, by age 4, they manifest no brain activity and remain in a vegetative state for 6 to 8 more years before eventual death. Such grim outcomes underscore the urgent need for the development of rational and effective therapeutic strategies, not only for INCL but also for all NCLs.
The aim of our clinical studies is to apply the knowledge gained from laboratory investigations to develop novel therapeutic strategies for Batten disease. The results of our earlier investigations on INCL (CLN1 disease) led to a bench-to-bedside clinical trial [Reference 1]. Using Cln1-knockout (Cln1–/–) mice, which recapitulate virtually all clinical and pathological features of INCL, we discovered that PPT1 deficiency causes endoplasmic-reticulum (ER) and oxidative stress, which at least in part causes neuronal death by apoptosis. During the past several years, we also delineated a mechanism by which PPT1 deficiency disrupts the recycling of the synaptic vesicle (SV) proteins, which are essential for regenerating fresh SVs to replenish the SV pool size at the nerve terminals to maintain uninterrupted neurotransmission. We also discovered that ER and oxidative stress contribute to neuronal apoptosis and neuro-inflammation in INCL. Further, we found that PPT1 deficiency causes misrouting of the V0a1 subunit of v-ATPase (the proton pump on lysosomal membrane), which regulates lysosomal acidic pH, causing elevated pH, which adversely affects lysosomal degradative function [Reference 2].
We also developed a non-invasive methods, using MRI and MRS (magnetic resonance spectroscopy), to evaluate the progression of neuro-degeneration in Ppt1–/– mice. The methods permit repeated evaluations of potential therapeutic agents in treated animals. Application of such methods in our clinical trial with INCL also allowed us to evaluate the progressive decline in brain volume and neuro-degeneration [Reference 3]. In collaboration with the NEI, we are also conducting studies to determine whether electro-retinography can be used to assess the progressive retinal deterioration in Cln1–/– as well as in the Cln1–knock-in (KI) mice generated in our laboratory, which carry the most common nonsense mutation found in the INCL patient population in the US. We also discovered that the blood-brain barrier is disrupted in Ppt1–/– mice and that the pathology is ameliorated by treatment with resveratrol, which has anti-oxidant properties. More recently, we discovered that a nucleophilic small molecule with antioxidant properties, N-(tert-butyl) hydroxylamine (NtBuHA), ameliorates the neurological abnormalities in Cln1–/– mice and extends their lifespan [Reference 4]. These and related studies provide insight into the complex mechanisms of heritable disorders of neuro-degeneration like INCL (CLN1 disease) and identify several potential therapeutic targets. Our results suggest that thioesterase-mimetic small molecules such as NtBuHA are potential therapeutic targets for INCL. More recently, we discovered that cathepsin D (CD) deficiency in lysosomes is a common pathogenic link between INCL (CLN1 disease) and congenital NCL (CNCL) or CLN10 disease. Our ongoing laboratory and clinical investigations are attempting to advance our knowledge of CLN1, CLN3, and CLN10 diseases. Our long-term plans are to apply the new findings arising from our laboratory studies to discover the pathogenic links among various NCLs and to develop novel therapeutic strategies not only for CLN1 disease but also for CLN3 and CLN10 diseases.
Lysosomal Ppt1 insufficiency may contribute to the pathogenesis of JNCL (CLN3-disease).
Even though mutations in at least 13 different genes cause various forms of NCLs, at the cellular level, all NCLs characteristically accumulate autofluorescent material (ceroid) in lysosomes and, clinically, patients develop seizures, visual failure, and experience a progressive decline in cognitive and motor functions. Such findings suggest shared pathogenic mechanism(s) among these diseases. It has been reported that CLN3 mutations suppress the exit of CI-M6PR from the trans-Golgi network (TGN). Notably, CI-M6PR transports soluble proteins like PPT1 from the TGN to the lysosome, although how this defect contributes to JNCL pathogenesis remains unclear. We showed that the lysosomes in the brain of Cln3–/– mice, which mimic JNCL, and those in cultured cells from JNCL patients contain significantly reduced levels of the Ppt1 protein and Ppt1 enzyme activity. Moreover, in Cln3–/– mice, lysosomal accumulation of S-palmitoylated proteins (constituents of ceroid) and a progressive increase in intracellular autofluorescence occur in an age-dependent manner. Furthermore, in Cln3–/– mice the V0a1 subunit of v-ATPase is mislocalized to the plasma membrane instead of its normal location on the lysosomal membrane, which elevates lysosomal pH, as previously reported in Cln1–/– mice, a reliable animal model of INCL [Reference 2]. We propose that lysosomal Ppt1 insufficiency is a pathogenic link between INCL and JNCL.
Altered lysosomal membrane localization of Rab7 dysregulates autophagy in a mouse model of INCL.
In eukaryotic cells, vesicular transport enables intracellular proteins to reach their destinations. In this process, a large superfamily of Ras-like GTPases (called Rabs) play pivotal roles in vesicle formation, cargo selection, sorting, transport, and vesicular fusion, which are all critical for endocytic and autophagic degradation. Rab7, one of the proteins belonging to the Rab superfamily of GTPases, directly or indirectly performs several important functions in the vesicular trafficking and membrane fusion events that occur between early endosome and late endosome/lysosome. Moreover, Rab7 (also known as RAB7A) facilitates the membrane fusion of endosome to lysosome and autophagosome to lysosome. The processes allow lysosomal acid hydrolases to encounter the cargo from both extracellular and intracellular sources that are delivered to the lysosome for degradation. Disruption of autophagosome-lysosome fusion is one of the suggested mechanisms for the accumulation of undegraded cargo in the lysosome leading to the LSDs.
As stated in the introduction, INCL is a devastating neurodegenerative LSD caused by mutations in the CLN1 gene encoding palmitoyl-protein thioesterase-1 (PPT1), which catalyzes depalmitoylation of S-palmitoylated proteins (constituents of ceroid) required for their degradation by lysosomal hydrolases. Thus, it is suggested that PPT1 deficiency causing lysosomal accumulation of ceroid leads to INCL. However, the molecular mechanism(s) of INCL pathogenesis remains poorly understood. Defective autophagy, due in part to impaired autophagosome-lysosome fusion, has been reported to underlie several LSDs, including some NCLs, but has not been reported in INCL. Even though impaired autophagosome-lysosome fusion is one of the suggested mechanism of dysfunctional autophagy, the precise mechanism of the defect remains unclear. A small GTPase, Rab7, plays critical roles in mediating autophagosome-lysosome fusion. We found that, in Cln1–/– mice, which mimic INCL, and in cultured cells from INCL patients, autophagy is dysregulated. We also demonstrate that Rab7 undergoes S-palmitoylation, a reversible post-translational modification of proteins by fatty acids (predominantly palmitate), which promotes protein stability and membrane anchorage. Moreover, dynamic S-palmitoylation (palmitoylation-depalmitoylation) facilitates the protein trafficking and steady-state membrane localization required for the function of many proteins. We found that dynamic S-palmitoylation of Rab7 is essential for its trafficking to the lysosomal membrane. Notably, Rab7 localization on lysosomal membrane is appreciably reduced in Ppt1–deficient Cln1–/– mice. Further, the interaction of Rab7 with its activator, RILP (Rab–Interacting Lysosomal Protein), which is required for its GTPase activity, was suppressed in Cln1–/– mice and in cultured INCL cells. The defect most likely impairs autophagosome-lysosome fusion, leading to intracellular accumulation of undegraded cargo (ceroid), thus contributing to INCL pathogenesis. Our findings uncover a previously unrecognized role of Cln1/Ppt1 in regulating Rab7 GTPase activity and suggest that suppression of autophagosome-lysosome fusion may contributes to INCL pathogenesis.
Dysregulation of lysosomal acidification in the INCL mouse model
In eukaryotic organisms, the lysosome is the primary organelle for intracellular digestion. It contains more than 50 hydrolases, which require an acidic pH for optimal degradative function. Thus, lysosomal acidification is of fundamental importance to the degradation of macromolecules of intra- and extracellular origin that are delivered to the lysosome. Moreover, it has been reported that dysregulation of lysosomal acidification contributes to pathogenesis in virtually all LSDs, including several NCLs. Furthermore, defective regulation of lysosomal pH has also been reported in common neuro-degenerative diseases such as Alzheimer’s and Parkinson’s disease. However, despite intense studies, the mechanism(s) underlying the lysosomal acidification defect remains largely unclear. Lysosomal acidification is regulated by vacuolar ATPase (v-ATPase), a multisubunit protein complex composed of the cytosolic V1 sector and the lysosomal membrane–anchored V0-sector. Reversible assembly of V1/V0 sectors on the lysosomal membrane maintains functionally active v-ATPase, the proton pump of the cell.
In the mammalian genome, 23 genes encode palmitoyl-acyl-transferases (PATs), which are evolutionarily conserved, cysteine-rich proteins containing Asp-His-His-Cys (DHHC) in the active site. In contrast, there are four thioesterases that have been characterized thus far. Two of these are cytosolic (acyl-protein thioesterase-1 [Apt1] and Apt2) and two (palmitoyl-protein thioesterase-1 [PPT1] and PPT2) are localized to the lysosome. Dynamic palmitoylation (palmitoylation-depalmitoylation), requiring coordinated action of both the DHHC-PATs and PPTs, maintains steady-state membrane localization and the function of numerous important proteins, especially in the brain. By catalyzing depalmitoylation, thioesterases also facilitate recycling or degradation of S-palmitoylated proteins (constituents of ceroid) by lysosomal hydrolases.
We tested the hypothesis that one or more subunits of v-ATPase requires S-palmitoylation for endosomal sorting, trafficking, and reversible assembly of V1/V0 on the lysosomal membrane, which is essential for regulating lysosomal pH, and that Ppt1 deficiency disrupts v-ATPase activity, impairing its proton transport function, thereby dysregulating acidification of lysosomal lumen. Our results show that the lysosomal membrane–anchored V0a1 subunit of v-ATPase undergoes dynamic S-palmitoylation, which is required for its sorting and trafficking to the lysosomal membrane [Reference 2]. The process appears to be defective in Ppt1–deficient Cln1–/– mice. Notably, we demonstrated that treatment of these mice with the thioesterase (Ppt1)–mimetic small molecule NtBuHA restores v-ATPase activity and rescues the defective lysosomal acidification phenotype.
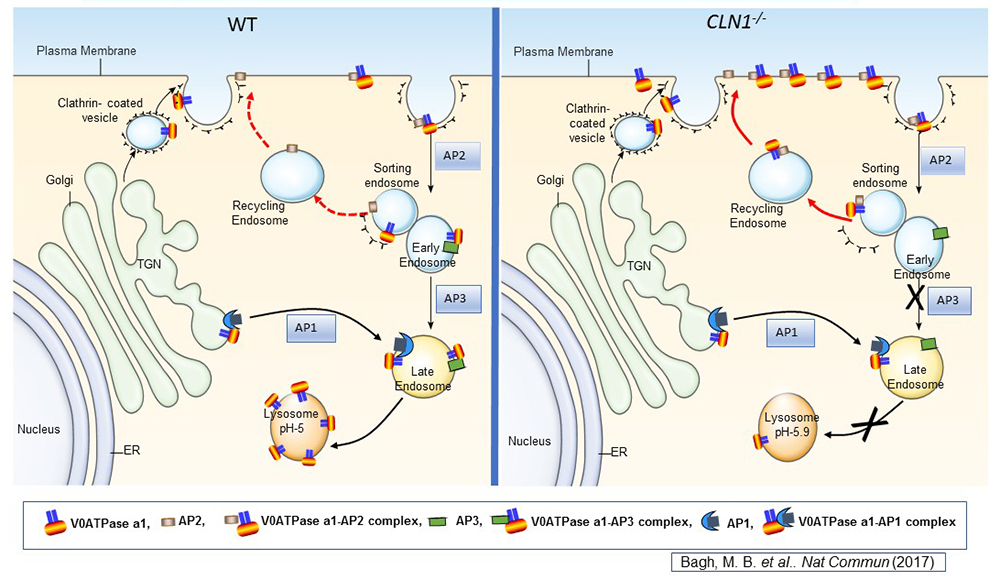
Click image to view.
Figure 1. Lysosomal acidification defect in a mouse model of infantile neuronal ceroid lipofuscinosis
Lysosomal acidification is accomplished by vacuolar ATPase (v-ATPase) localized to the lysosomal membrane. Consisting of a lysosomal membrane–localized V0 sector and cytosolic V1 sector, v-ATPase is a multi-subunit protein. We discovered that the V0a1 subunit of the v-ATPase requires S-palmitoylation (a reversible, post-translational modification of proteins by the 16-carbon saturated fatty acid palmitate) for trafficking from the trans-Golgi network (TGN) to the lysosomal membrane. The schematic representation shows the endosomal sorting and trafficking of V0a1 in wild-type (WT) and Cln1–/– mice, which mimic INCL. In Cln1–/– mice, as opposed to WT littermates, the V0a1 subunit of v-ATPase is misrouted to the plasma membrane instead of its normal location on the lysosomal membrane. The defect dysregulates lysosomal acidification. Given that lysosomal acid hydrolases require an acidic pH in the lysosomal lumen, elevated lysosomal pH contributes to diminished degradative function of these enzymes, thereby contributing to INCL pathogenesis.
Common pathogenic link between INCL (CLN1-disease) and CNCL (CLN10-disease)
The lysosome is the major degradative organelle responsible for disposing of the damaged macromolecules and organelles brought into the cell from external and internal sources. It has been reported that impaired lysosomal degradative capability leads to pathogenesis of many neurodegenerative disorders, including LSDs. Neurodegeneration is a manifestation in the majority of the more than 60 LSDs. Moreover, impaired lysosomal degradative capability has been reported in several late-onset neurodegenerative diseases such as Alzheimer’s, Huntington’s, and Parkinson’s disease. Cathepsin D (CD) is a major lysosomal aspartic protease essential for degradation of proteins delivered to the lysosome. Lysosomal CD activity catalyzes degradation and clearance of exogenous as well as endogenous macromolecules and damaged organelles delivered to the lysosome. Intracellular accumulation of undegraded long-lived proteins and other macromolecules leads to the pathogenesis of many neurodegenerative disorders. Paradoxically, both CD overexpression and CD deficiency have been reported to underlie neurodegenerative diseases. However, despite intense studies, this paradox has, until now, remained poorly understood.
Whereas inactivating mutations in the CLN1 gene, encoding palmitoyl-protein thioesterase-1 (PPT1), cause INCL, mutations in the CLN10/CTSD gene, encoding CD, underlie CNCL (CLN10-disease). We sought to determine whether there is a pathogenic link between INCL and CNCL. The synthesis of CD occurs in the endoplasmic reticulum (ER) as a pre-propeptide with a molecular mass of about 50 kDa. The cleavage of the leader peptide in the ER generates the 48 kDa precursor of mature CD (pro-CD). In the Golgi complex, attachment of mannose 6-phosphate to pro-CD facilitates the protein's binding to endosomal/lysosomal sorting receptors. The receptor-ligand complexes then exit the trans-Golgi network in clathrin-coated intermediates and fuse with the endosomal system. The low pH of the late endosomal lumen facilitates dissociation of the receptor-ligand complexes and allows the ligand (i.e., pro-CD) to be delivered to lysosome. The pro-CD then undergoes further proteolytic cleavage by cathepsin B (CB) and cathepsin L (CL), which generate, respectively, the 31 and 14 kDa fragments, non-covalent dimerization of which constitutes the mature, catalytically active CD. We used Cln1–/–/Ppt1–/– mice, which recapitulate virtually all clinical and pathological features of INCL, to test for a pathogenic link between INCL and CNCL. Our results show that, despite Cln10/Ctsd overexpression, defective processing of pro-CD to mature CD in lysosome leads to lysosomal CD deficiency, causing the neuropathology in INCL. Given that CD deficiency underlies CNCL, we propose that CD deficiency in the lysosome is indeed a common pathogenic link between INCL and CNCL.
Publications
- Levin SW, Baker EH, Zein WM, Zhang Z, Quezado ZM, Miao N, Gropman A, Griffin KJ, Bianconi S, Chandra G, Khan OI, Caruso RC, Liu A, Mukherjee AB. Oral cysteamine bitartrate and N-acetylcysteine for patients with infantile neuronal ceroid lipofuscinosis: a pilot study. Lancet Neurol 2014;13:777-787.
- Bagh MB, Peng S, Chandra G, Zhang Z, Singh SP, Pattabiraman N, Liu A, Mukherjee AB. Misrouting of v-ATPase subunit V0a1 dysregulates lysosomal acidification in a neurodegenerative lysosomal storage disease model. Nat Commun 2017;8:8:14612.
- Munasinghe J, Zhang Z, Kong E, Heffer A, Mukherjee AB. Evaluation of neurodegeneration in a mouse model of infantile batten disease by magnetic resonance imaging and magnetic resonance spectroscopy. Neurodegener Dis 2012;9(4):159-169.
- Sarkar C, Chandra G, Peng S, Zhang Z, Liu A, Mukherjee AB. Neuroprotection and lifespan extension in Ppt1(-/-) mice by NtBuHA: therapeutic implications for INCL. Nat Neurosci 2013;16:1608-1617.
- Chandra G, Bagh MB, Peng S, Saha A, Sarkar C, Moralle M, Zhang Z, Mukherjee AB. Cln1 gene disruption in mice reveals a common pathogenic link between two of the most lethal childhood neurodegenerative lysosomal storage disorders. Hum Mol Genet 2015;14:5416-5432.
Collaborators
- Eva Baker, MD, PhD, Radiology and Imaging Sciences, Clinical Center, NIH, Bethesda, MD
- Yichao Li, Visual Function Core, NEI, Bethesda, MD
- Chris J. McBain, PhD, Section on Cellular and Synaptic Physiology, NICHD, Bethesda, MD
- Kenneth Pelkey, PhD, Section on Cellular and Synaptic Physiology, NICHD, Bethesda, MD
- Haohua Qian, PhD, Visual Function Core, NEI, Bethesda, MD
- Ling-Gang Wu, PhD, Synaptic Transmission Section, NINDS, Bethesda, MD
- Wadih M. Zein, MD, Ophthalmic Genetics and Visual Function Branch, NEI, Bethesda, MD
Contact
For more information, email mukherja@exchange.nih.gov or visit irp.nih.gov/pi/anil-mukherjee.