Membrane Rearrangements in Viral Entry and Developmental Fusion

- Leonid V. Chernomordik, PhD, Head, Section on Membrane Biology
- Eugenia Leikina, DVM, Senior Research Assistant
- Kamram Melikov, PhD, Staff Scientist
- Elena Zaitseva, PhD, Staff Scientist
- Santosh K. Verma, PhD, Visiting Fellow
- Berna Uygur, PhD, Postdoctoral Intramural Research Training Award Fellow
- Kepler Mears, BS, Postbaccalaureate Fellow
Diverse biological processes, in which enveloped viruses infect cells and cells from all kingdoms of life secrete, internalize, traffic and sort integral proteins, sculpt their membranes, and bring together parent genomes in sexual reproduction, share a common stage: fusion of two membranes into one. Biological membrane remodeling is tightly controlled by protein machinery but is also dependent on the lipid composition of the membranes. Whereas each kind of protein has its own, individual personality, membrane lipid bilayers have rather general properties, manifested by their resistance to disruption and bending, and by their charge. Our long-term goal is to understand how proteins fuse membrane lipid bilayers. We expect that better understanding of important fusion reactions will bring about new ways of controlling them and will lead to new strategies for quelling diseases involving cell invasion by enveloped viruses and defects in intracellular trafficking or intercellular fusion. Our general strategy is to combine in-depth analysis of the best characterized fusion reactions with comparative analysis of diverse, less explored fusion reactions, which can reveal new kinds of fusion proteins and clarify the generality of emerging mechanistic insights. In our recent studies, we explored mechanisms of the myoblast fusion stage of development, of the regeneration of skeletal muscles, and of the osteoclast precursor fusion stage during the formation of multinucleated osteoclasts.
Distinct functions of myomaker and myomerger during myoblast fusion
Multinucleated skeletal muscle fibers form through the fusion of myoblasts during development and regeneration. Previous studies identified myomaker (Tmem8c) and myomerger as muscle-specific membrane proteins essential for fusion [Millay DP, et al. Nature 2013;499:301; Quinn ME, et al. Nat Commun 2017;8:15665]. In our recent study (collaboration with Douglas Millay) we explored the localization of endogenous myomaker in muscle cells [Reference 1]. We found myomaker at the plasma membrane and also in the Golgi and post-Golgi vesicles. Trafficking of myomaker is regulated by palmitoylation of C-terminal cysteine residues, which allows Golgi localization. To examine whether myomaker functions at the myoblast fusion stage or only prepares the cells for fusion, we applied a synchronized fusion approach developed in earlier studies of our lab [Leikina E, et al. J Cell Biol 2013;200:109]. We reversibly blocked fusion of myoblasts without blocking pre-fusion stages using lysophosphatidylcholine (LPC), an inhibitor of membrane merger. We accumulated ready-to-fuse cells in the presence of LPC and then removed LPC to allow fusion. Myomaker antibody applied at the time of LPC removal blocked lipid mixing and content mixing, indicating that myomaker at the plasma membrane at the time and place of fusion is directly involved in the early stages of myoblast fusion.
In our most recent report [Reference 2], we furthered the understanding of myoblast fusion by demonstrating that the fusion reaction proceeds through a novel step-wise, bi-factorial mechanism whereby different proteins function at independent points in the pathway. We demonstrated that the myoblast fusion reaction proceeds through a novel step-wise, bi-factorial mechanism whereby different proteins function at independent points in the pathway [Reference 2]. While myomaker function is essential for fusion initiation and hemifusion, myomerger drives subsequent fusion pore formation (Figure 1). As reported in our earlier work, fusion pore formation and expansion also depend on dynamin, cell metabolism, and phosphatidylinositol 4,5-bisphosphate [Leikina E, et al. J Cell Biol 2013;200:109]. In better characterized fusion systems (viral and intracellular membrane fusion), reactions can be stalled at the hemifusion stage, but both hemifusion and pore formation are driven by the same protein complex. Thus, we revealed a membrane fusion mechanism that is divergent from well established models of fusion such as SNARE–mediated intracellular fusion, hemagglutinin (HA)–mediated viral fusion, and developmental fusion mediated by the cell fusogen Eff-1. Myomaker does not require myomerger to mediate hemifusion, as evidenced by the finding that the treatments promoting hemifusion-to-fusion transition of myomerger–/– myoblasts result in levels of complete fusion comparable to wild-type (WT) levels, showing that the lack of myomerger does not impact myomaker-dependent hemifusion. Similarly, myomerger does require myomaker to drive the hemifusion-to-fusion transition, as evidenced by the finding that myomerger drives completion of heterologous fusion reaction mediated by the viral fusion protein HA, a system completely devoid of myomaker. In summary, we distinguished the functions served by single fusogens in other systems and assigned to two independently acting muscle-specific proteins that function to drive distinct fusion stages. Our results suggest that the division of the reaction could be a regulatory checkpoint for myoblast fusion. A deeper understanding of the myoblast fusion mechanism could help develop new therapeutic strategies for genetic and acquired muscle diseases.
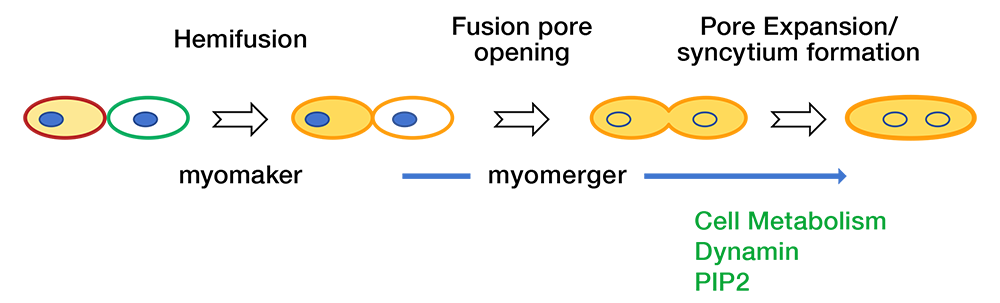
Click image to view.
Figure 1. Schematic of the division of fusogenic labor between myomaker and myomerger
Myomaker is indispensable for hemifusion, and myomerger is not required for this step. The fusion reaction would stall at this stage without the presence of myomerger, which at the second stage of the reaction drives opening of a fusion pore [Reference 2]. A subsequent expansion of the pore also depends on dynamin, cell metabolism and PIP2 [Leikina E, et al. J Cell Biol 2013;200:109]. Other factors that may be important for fusion (for instance annexin A5 [Leikina E, et al. J Cell Biol 2013;200:109]) are omitted for clarity.
Phosphatidylserine at the surface of osteoclast precursors regulates their fusion.
In our recent study [Reference 4], we focused on the cell-cell fusion stage of osteoclast formation. Multinucleated osteoclasts resorb bones to balance the bone-forming activity of osteoblasts in the continuous bone-remodeling process in both healthy animals and in pathological states. Osteoclasts are formed from precursor cells (OCPs) of monocyte/macrophage lineage in the presence of macrophage colony stimulating factor (M-CSF) and receptor activator of NF-kappa-B ligand (RANKL). Many groups have characterized the osteoclastogenesis using in vitro models based on human monocytes (HMs), murine bone marrow cells (BMC), and macrophage-like murine monocytic RAW 264.7 cells (“RAW cells”). Several proteins, including: a regulator of immune properties of dendritic cells, the dendritic cell–specific transmembrane protein DC-STAMP; the osteoclast-stimulatory transmembrane protein (OC-STAMP); purinergic receptors; S100 proteins; protein tyrosine phosphatase PEST; an adaptor protein Tks5; an intermediate-conductance calcium-activated potassium channel; and CD47, were shown to be involved in osteoclastogenesis, and it has been suggested that they are also involved in OCP fusion. Recent studies also demonstrated that the formation of multinucleated osteoclasts depends on clathrin-mediated endocytosis. The specific stages of osteoclastogenesis that are dependent on the proteins listed above (fusion vs. pre- or post-fusion stages) remain to be clarified. Generation of multinucleated osteoclasts also involves syncytin-1 (Syn-1), the envelope protein of a human endogenous retrovirus, HERVW1. Syn-1 is highly expressed in placental trophoblasts and mediates their fusion in human placentogenesis. Fusogenic activity of Syn-1 is triggered by its interactions with ASCT1/2 receptors. Suppression of Syn-1 activity inhibits both formation of multinucleated human osteoclasts and expression of a biochemical marker of osteoclast maturation, tartrate-resistant acidic phosphatase (TRAP). Given that TRAP expression develops independently of cell–cell fusion, these findings suggest that Syn-1 either functions in both the fusion stage and the pre-fusion stages.
We uncoupled the cell-fusion step from both pre-fusion stages of osteoclastogenic differentiation and the post-fusion expansion of the nascent fusion connections [Reference 4]. As for myoblast fusion [Leikina E, et al. J Cell Biol 2013;200:109] and in our earlier work on osteoclast fusion [Verma SK, Leikina E, Melikov K, Chernomordik LV. Biochem J 2014;464:293], we accumulated ready-to-fuse cells in the presence of LPC and then removed it to study synchronized cell fusion. We found that osteoclast fusion requires the DC-STAMP–dependent non-apoptotic exposure of phosphatidylserine at the surface of fusion-committed cells. Fusion also depended on annexins, extracellular phosphatidylserine binding proteins (annexin A5 in human osteoclasts), which, along with annexin-binding protein S100A4, regulated fusogenic activity of syncytin 1. Thus, in contrast to fusion processes mediated by a single protein, such as epithelial cell fusion in C. elegans, cell fusion step in osteoclastogenesis is controlled by the phosphatidylserine-coordinated activity of several proteins.
In addition to identification of protein and lipid players in osteoclast fusion, in our recent studies we suggested new ways of unbiased presentation of cell fusion at given conditions that combine the empirical cumulative distribution function for the sizes of multinucleated cells with the total number of cell-cell fusion events, which generate these cells [References 3, 4].
Additional Funding
- United States-Israel Binational Science Foundation (BSF) grant “Machinery of myoblast fusion” 2015-2019
- Office of Aids Research Award, 2017, 2018
- NICHD Director's Awards, 2018, 2019
- NICHD Zika Research Award, 2018
Publications
- Gamage DG, Leikina E, Quinn ME, Ratinov A, Chernomordik LV, Millay DP. Insights into the localization and function of myomaker during myoblast fusion. J Biol Chem 2017;292(42):17272-17289.
- Leikina E, Gamage DG, Prasad V, Goykhberg J, Crowe M, Diao J, Kozlov MM, Chernomordik LV, Millay D. Myomaker and Myomerger work independently to control distinct steps of membrane remodeling during myoblast fusion. Dev Cell 2018;46:767-780.
- Verma SK, Chernomordik LV, Melikov K. An improved metrics for osteoclast multinucleation. Sci Rep 2018;8:1768.
- Verma SK, Leikina E, Melikov K, Gebert C, Kram V, Young MF, Uygur B, Chernomordik LV. Cell-surface phosphatidylserine regulates osteoclast precursor fusion. J Biol Chem 2018;293:254-270.
Collaborators
- Anush Arakelyan, PhD, Section on Intercellular Interactions, NICHD, Bethesda, MD
- Claudia M. Gebert, PhD, Section on Genome Imprinting, NICHD, Bethesda, MD
- Michael M. Kozlov, PhD, Dhabil, Sackler Faculty of Medicine, Tel Aviv University, Tel Aviv, Israel
- Leonid Margolis, PhD, Section on Intercellular Interactions, NICHD, Bethesda, MD
- Douglas Millay, PhD, Cincinnati Children's Hospital Medical Center, Cincinnati, OH
- Benjamin Podbilewicz, PhD, Technion-Israel Institute of Technology, Haifa, Israel
- Marian F. Young, PhD, Molecular Biology of Bones and Teeth Section, NIDCR, Bethesda, MD
Contact
For more information, email chernoml@mail.nih.gov or visit www.nichd.nih.gov/research/atNICHD/Investigators/chernomordik.