Diagnosis, Localization, Pathophysiology, and Molecular Biology of Pheochromocytoma and Paraganglioma

- Karel Pacak, MD, PhD, DSc, Head, Section on Medical Neuroendocrinology
- Thanh-Truc Huynh, MS, Biologist
- Karen T. Adams, MSc, CRNP, Research Nurse
- Tamara Prodanov, MD, Clinical Trial Database (CTDB) Coordinator
- Suman Goshal, PhD, Postdoctoral Visiting Fellow
- Abishek Jha, MBBS, Postdoctoral Visiting Fellow
- Liping Li, MD, PhD, Postdoctoral Visiting Fellow
- Ying Pang, MD, PHD, Postdoctoral Visiting Fellow
- Alrezk Rami, MD, PhD, Clinical Fellow
- Veronika Caisova, MS, Predoctoral Visiting Fellow
- Melissa Gonzales, BS, Postbaccalaureate Fellow
- Neil F. Amba, MD, Volunteer
- Maria Gungon, MD, Volunteer
- Marcela Komrskova, DPharm, Volunteer
- Geline Malaza, MD, Volunteer
- Divya Mamilla, MD, Volunteer
- Cecilia Angela Paraiso, MD, PhD, Volunteer
- Mayank Patel, MD, Volunteer
- Ashraf Shoaib, MD, Volunteer
- Andres Suarez Parraga, MD, Volunteer
- Isabel Tena Garcia, MD, Volunteer
- Marianne Knue, CRNP, Nurse Practitioner
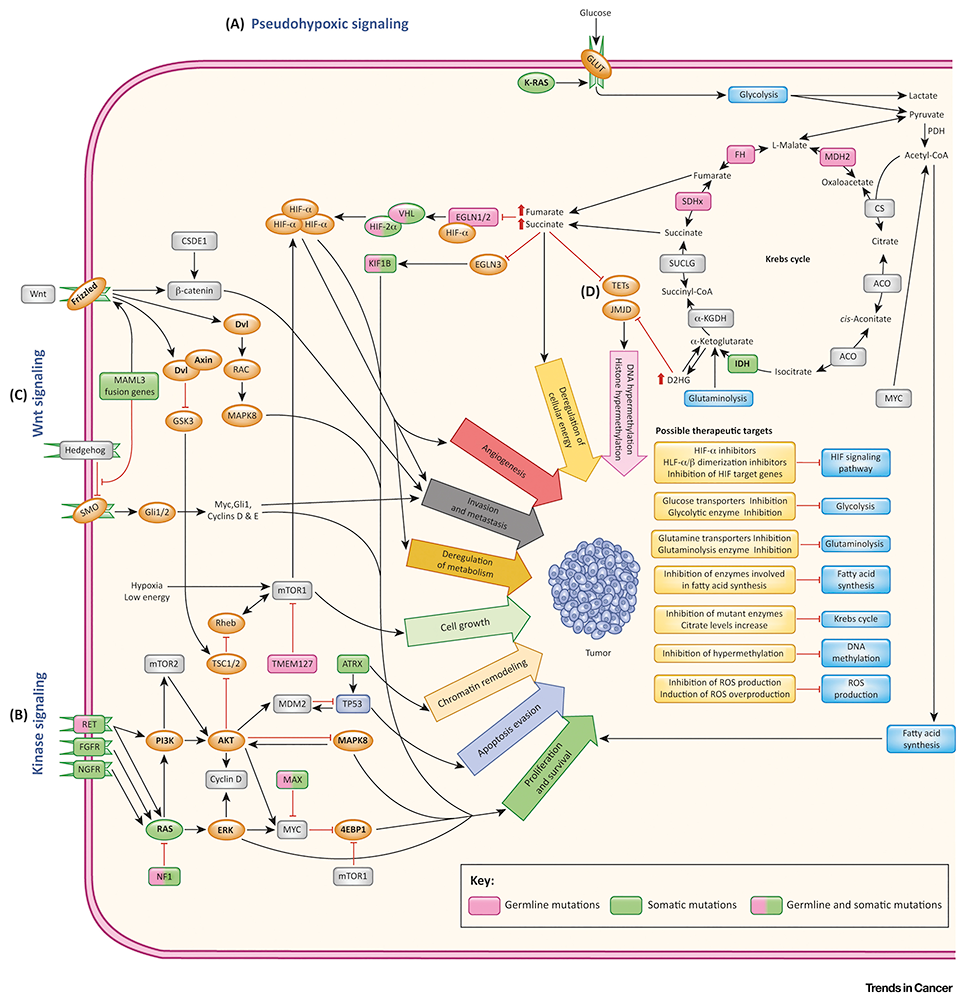
Pheochromocytomas (PHEOs) and paragangliomas (PGLs) are rare and clinically important chromaffin cell tumors that typically arise, respectively, from the adrenal gland and from extra-adrenal paraganglia. The clinical features and consequences of PHEO/PGL result from the release of catecholamines (norepinephrine and epinephrine). An undetected PHEO/PGL poses a hazard to patients undergoing surgery, childbirth, or general anesthesia, because of the potential for excess catecholamine secretion, which can result in significant, often catastrophic outcomes. Diagnosing and localizing a PHEO/PGL can be challenging. Plasma and urinary catecholamines as well as their metabolites and radio-iodinated metaiodobenzylguanidine (MIBG) scanning can yield false-positive/negative results in patients harboring the tumor, and computed tomography (CT) and magnetic resonance imaging (MRI) lack sufficient specificity. The molecular mechanisms by which genotypic changes predispose to the development of PHEO/PGL remain unknown, even in patients with identified mutations. Moreover, in patients with hereditary predispositions, PHEOs/PGLs differ in terms of their growth, malignant potential, catecholamine phenotype, responses to standard screening tests, various imaging modalities, and therefore, subsequently, to different therapeutic options. Our studies focuses on developmental, molecular, genetic, epigenetic, proteomic, metabolomic, and other types of studies to investigate the bases for predisposition to develop PHEOs/PGLs and the expression of different neurochemical phenotypes and malignant potentials, including therapeutic responses.
Click image to view.
Figure 1. Animal model of pheochromocytoma and cell culture studies
Ongoing studies are focusing on the development of SDHB– and HIF2A–related PHEO/PGL. We also started working on animal experiments using innate immunity approaches to treat subcutaneous and on metastatic PHEO in mice.
Clinical and genetic aspects of pheochromocytoma and paraganglioma
Germline or somatic mutations of numerous genes have been implicated in the pathogenesis of pheochromocytomas/paragangliomas (PPGLs), including the isocitrate dehydrogenase 1 (IDH1) gene and alpha thalassemia/mental retardation syndrome X-linked (ATRX) gene. Although concurrent IDH1 and ATRX mutations are frequently seen in gliomas, they have never been reported together in PPGLs. The aim of this study was to characterize one paraganglioma with concurrent IDH1 and ATRX mutations identified by whole exome sequencing. We used leukocyte and tumor DNA for whole exome sequencing and Sanger sequencing. We measured the 2-hydroxyglurarate level and the global DNA methylation status in the tumor and analyzed ATRX's cDNA transcripts. Tyrosine hydroxylase (TH), HIF1α and ATRX staining, as well as telomere-specific FISH were also performed. In this particular case, we confirmed the presence of a somatic IDH1 (c.394C→T, p.R132C) mutation and a concurrent somatic ATRX splicing mutation (c.4318-2A→G). Dramatic accumulation of 2-hydroxyglutarate was detected in the paraganglioma without the global DNA hypermethylation, and pseudohypoxia was also activated. Importantly, immunohistochemistry revealed negative TH staining in the tumor, and the first exon region of the TH gene was hypermethylated, resulting in normal plasma metanephrines. The splicing ATRX mutation resulted in two transcripts, causing frameshifts. Immunohistochemistry revealed scarce ATRX staining in the tumor. Alternative lengthening of telomeres (ALT) was detected by FISH. The case represented the first concurrence of IDH1 and ATRX mutations in PPGLs. Although relatively rare, a somatic R132C mutation of IDH1 might play a role in a small subset of sporadic PPGLs.
Our knowledge of the susceptibility genes for PPGLs has widened; however, data on its impact on surgical decision-making has not been described. The aim of this study was to determine the effect of routine preoperative genetic testing on the operative intervention in patients with PPGLs. We subjected 108 patients diagnosed with PPGLs, who underwent 118 operations, to preoperative genetic testing for 9 known PPGL susceptibility genes and performed a retrospective analysis of a prospective database to evaluate clinical factors associated with the surgical approach selected and the outcome of the surgical intervention. In 51 patients (47%), a germline mutation was detected and one-third had no family history of PPGL. In 77 operations (65%), it was the first operative intervention for the disease site (60 laparoscopic, 17 open), and 41 (35%) were reoperative interventions (36 open, 5 laparoscopic). For initial operations, variables associated with whether an open or laparoscopic approach was used were tumor size and the presence of germline mutation. Sixty-eight adrenal operations were performed (54 total, 14 cortical-sparing). Variables significantly associated with a cortical-sparing adrenalectomy being performed were the presence of germline mutation and tumor size. We concluded, that preoperative knowledge of the germline mutation status affects the surgical approach and extent of adrenalectomy.
We also provided the first direct molecular-genetic evidence of an association between a somatic iron-regulatory protein 1 (IRP1) loss-of-function mutation and pheochromocytoma and secondary polycythemia. In patients diagnosed with PPGL and polycythemia with negative genetic testing for mutations in HIF2A (gene encoding hypoxia-inducible factor alpha), PHD1/2 (gene encoding hypoxia-inducible factor prolyl hydroxylase 1), and VHL (Von Hippel–Lindau disease tumor suppressor gene), IRP1 should be considered a candidate gene.
Imaging of pheochromocytomas and paragangliomas
We evaluated and compared the diagnostic performance of 68Ga-DOTA(0)-Tyr(3)-octreotate (68Ga-DOTATATE) with 18F-fluoro-2-deoxy-¿-glucose (18F-FDG) positron emission tomography–computed tomography (PET/CT) and with anatomic imaging using computed tomography and/or magnetic resonance (CT/MR) imaging in the detection of mutated succinate dehydrogenase (SDHx)–related PPGLs in pediatric patients. Nine pediatric patients (5:4, girls:boys; 14.6 ± 2.0 years) with an SDHx–related mutation (SDHB:SDHA:SDHD, n = 7:1:1) were included in this retrospective study. At the time of initial diagnosis, 7 of 9 patients had metastatic disease. They all underwent CT/MR imaging along with PET/CT using 68Ga-DOTATATE (n = 9), 18F-FDG (n = 8), and positron emission tomography–magnetic resonance imaging (PET/MR) using 18F-FDG (n = 1). We compared the per-lesion, per-region, and per-patient detection rates and calculated them for each of the imaging modalities. A composite of all functional and anatomic imaging studies served as the imaging comparator. Eight out of nine patients were positive for PPGLs on the imaging studies, which demonstrated 107 lesions in 22 anatomic regions on the imaging comparator. The per-lesion detection rates for 68Ga-DOTATATE PET/CT, 18F-FDG PET/CT (18F-FDG PET/CT refers to both 18F-FDG PET/CT and 18F-FDG PET/MR), and CT/MR imaging were 93.5% (95% CI: 87.0–97.3%), 79.4% (95% CI: 70.5–86.6%), and 73.8% (95% CI: 64.5–81.9%), respectively. The per-lesion detection rate for 68Ga-DOTATATE PET/CT was significantly higher than that for 18F-FDG PET/CT or for CT/MR imaging. In all anatomic regions except the abdomen, the per-lesion detection rates for 68Ga-DOTATATE PET/CT was found to be equal or superior to 18F-FDG PET/CT, and CT/MR imaging. The per-region detection rate was 100% (95% CI: 84.6–100%) for 68Ga-DOTATATE PET/CT and 90.9% (95% CI: 70.8–98.9%) for both 18F-FDG PET/CT and CT/MR imaging. The per-patient detection rates for 68Ga-DOTATATE PET/CT, 18FDG PET/CT, and CT/MR imaging were all 100% (95% CI: 63.1–100%). We thus demonstrated the superiority of 68Ga-DOTATATE PET/CT in localization of SDHx–related PPGLs in pediatric population over 18F-FDG PET/CT and CT/MR imaging with the exception of abdominal (excluding adrenal and liver) lesions, which suggests that it might be considered as a first-line imaging modality in pediatric patients with SDHx–related PPGLs.
MYC–associated factor X (MAX) has been recently described as a new susceptibility pheochromocytoma (PHEO) gene with a total of about 40 reported cases. At present, no study has specifically described the functional imaging phenotype of MAX–related pheochromocytoma. We reported our experience with contrast-enhanced computed tomography (CT) and 18F-fluorodihydroxyphenylalanine (18F-FDOPA) positron emission tomography (PET)/CT in six consecutive patients (four at the initial diagnosis and two at the follow-up evaluation) with rare, but clinically important, MAX–related pheochromocytomas. In five patients, we compared 18F-FDOPA with other radiopharmaceutical agents. The patients had five different mutations in the MAX gene that caused disruption of Max/Myc interaction and/or abolished interaction with DNA, as determined by in silico analyses. All but one patient developed bilateral pheochromocytomas during their lifetime. In all cases, 18F-FDOPA PET/CT accurately visualized pheochromocytomas, which were often multiple within the same gland or bilaterally, and detected more adrenal and extra-adrenal lesions than did CT (per-lesion sensitivity, 90.9% vs 52.4% for CT/magnetic resonance imaging). The two pheochromocytomas missed on 18F-FDOPA PET/CT were less than 1 cm in size, corresponding to nodular adrenomedullary hyperplasia. 68Ga-DOTA,Tyr3-octreotate PET/CT detected fewer lesions than did 18F-FDOPA PET/CT in one of three patients, and 18F-FDG PET/CT was only faintly positive in two of four patients, with underestimation of extra-adrenal lesions in one patient. We concluded that MAX–related pheochromocytomas exhibit a marked 18F-FDOPA uptake, a finding that illustrates the common well differentiated chromaffin pattern of pheochromocytomas associated with activation of kinase signaling pathways. 18F-FDOPA PET/CT should be considered as the first-line functional imaging modality for diagnostic or follow-up evaluations for these patients.
In recent years, inherited and acquired mutations in genes encoding the tricarboxylic acid (TCA) cycle enzymes have been reported in diverse cancers. PPGLs often exhibit dysregulation of glucose metabolism, which is also driven by mutations in genes encoding the TCA cycle enzymes or by activation of hypoxia signaling. PPGLs associated with SDH deficiency are characterized by high 18F-FDG avidity, an association that is currently only partially understood. We therefore hypothesized that accumulation of succinate resulting from the TCA–cycle defect could be the major connecting hub between SDH–mutated tumors and the 18F-FDG uptake profile. To test whether succinate modifies the 18F-FDG metabolic profile of tumors, we performed in vitro and in vivo (small-animal PET/CT imaging and autoradiography) experiments in the presence of succinate, fumarate, and phosphate-buffered saline (PBS) in various cell models. As a control, we also evaluated the impact of succinate on 18F-fluorocholine uptake and retention. We performed glucose transporter 1 (GLUT1) immunohistochemistry to assess whether 18F-FDG uptake correlates with GLUT1 staining. Intra-tumoral injection of succinate significantly increased 18F-FDG uptake at 24 h on small-animal PET/CT imaging and autoradiography. No effect of succinate was observed on cancer cells in vitro, but interestingly we found that succinate elevated 18F-FDG uptake by human umbilical vein endothelial cells in a concentration-dependent manner. No significant effect was observed after intra-tumoral injection of fumarate or PBS. Succinate, fumarate, and PBS have no effect on cell viability, regardless of cell lineage. Intramuscular injection of succinate also significantly increases 18F-FDG uptake by muscle when compared with either PBS or fumarate, highlighting the effect of succinate on connective tissues. We observed no difference between PBS and succinate on 18F-fluorocholine uptake in the tumor or muscle or in hind limb blood flow. GLUT1 expression quantification did not significantly differ between the study groups. We showed that succinate stimulates 18F-FDG uptake by endothelial cells, a finding that partially explains the 18F-FDG metabotype observed in tumors with SDH deficiency. Although this study is an 18F-FDG–based approach, it provides an impetus to better characterize the determinants of 18F-FDG uptake in various tumors and their surrounding microenvironment, with special emphasis on the role of tumor-specific oncometabolites.
Therapeutic aspects of pheochromocytoma and paraganglioma
Patients harboring germline mutations in SDHB present with PPGLs that are more likely to be malignant and clinically aggressive. We retrospectively evaluated the combination chemotherapy of cyclophosphamide, vincristine, and dacarbazine (CVD) in patients with SDHB-associated metastatic PPGL. Twelve metastatic PPGL patients harboring SDHB mutations/polymorphisms with undetectable SDHB immunostaining were treated with CVD. CVD therapy consisted of 750 mg/m2 cyclophosphamide with 1.4 mg/m2 vincristine on day 1 and 600 mg/m2 dacarbazine on days 1 and 2, every 21–28 days. We determined treatment outcome by RECIST (response evaluation criteria in solid tumors) criteria as by response duration and progression-free and overall survivals. A median of 20.5 cycles (range 4–41) was administered. All patients experienced tumor reduction (12–100% by RECIST). Complete response was seen in two patients, while partial response was observed in 8. The median number of cycles to response was 5.5. Median duration of response was 478 days, with progression-free and overall survivals of 930 and 1190 days, respectively. Serial [18F]-FDG PET and CT imaging demonstrated continued incremental reduction in maximal standardized uptake values (SUVmax) values in 26/30 lesions. During treatment administration, the median SUV decreased from over 25 to less than 6, indicating the efficacy of chemotherapy over a prolonged period of time. Prolonged therapy results in continued incremental tumor reduction and is consistent with persistent drug sensitivity. CVD chemotherapy is recommended as part of the initial management in patients with metastatic SDHB–related PPGL.
Metastatic PPGLs are frequently associated with SDHB mutations. The CVD regimen is recommended as standard chemotherapy for advanced mPPGL. There is limited evidence to support the role of metronomic schemes (MS) of chemotherapy in metastatic PHEO/PGL treatment. We report two patients with SDHB–related metastatic PGL who received a regimen consisting of MS temozolomide (TMZ) and high-dose lanreotide after progression on both CVD chemotherapy and high-dose lanreotide. Molecular profiling of the tumor tissue from both patients revealed hypermethylation of the O-6-methylguanine-DNA–methyltransferase (MGMT) promoter (MGMT is a DNA repair enzyme). In one patient, progression-free survival was 13 months and the second patient remained under treatment after 27 months of stabilization of the metabolic response of his disease. Treatment was well tolerated, and adverse effects were virtually absent. A modification in the scheme of TMZ from standard schemes to MS is safe and feasible and can be considered in patients with progressive metastatic PPGL refractory to dacarbazine in standard doses.
Cluster I PPGLs tend to develop malignant transformation, tumor recurrence, and multiplicity. Transcriptomic profiling suggests that cluster I PCPGs and other related tumors exhibit distinctive changes in the TCA cycle (the central driver of cellular respiration), the hypoxia signaling pathway, the mitochondrial electron transport chain, and methylation status, suggesting that the therapeutic regimen might be optimized by targeting these signature molecular pathways. We thus investigated the molecular signatures in clinical specimens from cluster I PPGLs in comparison with cluster II PPGLs that are related to kinase signaling and often present as benign tumors. We found that cluster I PPGLs develop a dependency to mitochondrial complex I, evidenced by the upregulation of complex I components and enhanced NADH dehydrogenation. Alteration in mitochondrial function resulted in strengthened NAD+ metabolism, here considered a key mechanism of chemoresistance, in particular of succinate dehydrogenase subunit B (SDHB)–mutated cluster I PPGLs via the PARP1/BER DNA repair pathway. Combining a PARP inhibitor with temozolomide, a conventional chemotherapeutic agent, not only improved cytotoxicity but also reduced metastatic lesions, with prolonged overall survival of mice with SDHB knockdown PPGLs allograft. Our findings provided novel insights into an effective strategy for targeting.
Proteasome inhibitors have been used frequently in treating hematologic and solid tumors. They are administered individually or in combination with other regimens, to prevent severe side effects and resistance development. Because they have been shown to be efficient and are pharmaceutically available, we tested the first FDA–approved proteasome inhibitor bortezomib alone and in combination with another proteasome inhibitor, salinosporamid A, in pheochromocytoma cells. Given that PPGLs are neuroendocrine tumors for which no definite cure is yet available, drugs with a wide spectrum of mechanisms of action are being tested to identify suitable candidates for PPGL treatment. We showed that bortezomib induces pheochromocytoma cell death via the apoptotic pathway both in vitro and in vivo. The combination of bortezomib with salinosporamid A exhibits additive effect on these cells and inhibits proliferation, cell migration and invasion, and angiogenesis more potently than bortezomib alone. Altogether, we suggest the proteasome inhibitors, especially bortezomib, could be tested in PPGL patients who might benefit from treatment with either the inhibitors alone or in combination with other treatment options.
Metabolic aspects of PHEO/PGL
Metabolic aberrations have been described in neoplasms with pathogenic variants in the Krebs cycle genes encoding SDH, fumarate hydratase (FH), and isocitrate dehydrogenase (IDH). In turn, accumulation of the oncometabolites succinate, fumarate, and 2-hydroxyglutarate can be employed to identify tumors with those pathogenic variants. Additionally, such metabolic readouts may aid in genetic variant interpretation and improve diagnostics. Using liquid chromatography–mass spectrometry, 395 PPGLs from 391 patients were screened for metabolites to indicate Krebs cycle aberrations. We used multigene panel sequencing to detect driver pathogenic variants in cases with indicative metabolite profiles but undetermined genetic drivers. Aberrant Krebs cycle metabolomes identified rare cases of PPGLs with germline pathogenic variants in FH and somatic pathogenic variants in IDH and SDH, including the first case of a somatic IDH2 pathogenic variants in PPGL. Metabolomics also reliably identified PPGLs with SDH loss-of-function (LoF) pathogenic variants. Therefore, we utilized tumor metabolite profiles to further classify variants of unknown significance in SDH, thereby enabling missense variants associated with SDH LoF to be distinguished from benign variants. We proposed incorporation of metabolome data into the diagnostics algorithm in PPGLs to guide genetic testing and variant interpretation and to help identify rare cases with pathogenic variants in FH and IDH.
Animal model of pheochromocytoma and cell culture studies
Ongoing studies are focusing on the development of SDHB– and HIF2A–related PHEO/PGL. We also started working on animal experiments using innate immunity approaches to treat subcutaneous and on metastatic PHEO in mice.
Publications
- Crona J, Taïeb D, Pacak K. New perspectives on pheochromocytoma and paraganglioma: towards a molecular classification. Endocr Rev 2017;38(6):489-515.
- Taïeb D, Pacak K. New insights into the nuclear imaging phenotypes of cluster 1 pheochromocytoma and paraganglioma. Trends Endocrinol Metab 2017;28(11):807-817.
- Jha A, Ling A, Millo C, Gupta G, Viana B, Lin FI, Herscovitch P, Adams KT, Taïeb D, Metwalli AR, Linehan WM, Brofferio A, Stratakis CA, Kebebew E, Lodish M, Civelek AC, Pacak K. Superiority of 68Ga-DOTATATE over 18F-FDG and anatomic imaging in the detection of succinate dehydrogenase mutation (SDHx)-related pheochromocytoma and paraganglioma in the pediatric population. Eur J Nucl Med Mol Imaging 2018;45(5):787-797.
- Pang Y, Lu Y, Caisova V, Liu Y, Bullova P, Huynh TT, Zhou Y, Yu D, Frysak Z, Hartmann I, Taieb D, Pacak K, Yang C. Targeting NAD+/PARP DNA repair pathway as a novel therapeutic approach to SDHB-mutated cluster I pheochromocytoma and paraganglioma. Clin Cancer Res 2018;24(14):3423-3432.
- El Lakis M, Gianakou A, Nockel P, Wiseman D, Tirosh A, Quezado MA, Patel D, Nilubol N, Pacak K, Sadowski SM, Kebebew E. Radioguided surgery with gallium 68 dotatate for patients with neuroendocrine tumors. JAMA Surg 2019;154(1):40-45.
Collaborators
- James Bibb, PhD, University of Alabama Comprehensive Cancer Center, University of Alabama at Birmingham Medical Center, Birmingham, AL
- Clara C. Chen, MD, Nuclear Medicine Department, Clinical Center, NIH, Bethesda, MD
- Hans Clevers, PhD, Hubrecht Institute, Royal Netherlands Academy of Sciences, Utrecht, The Netherlands
- Peter Deen, PhD, Radboud Institute for Molecular Life Sciences, Nijmegen, The Netherlands
- Graeme Eisenhofer, PhD, Universität Dresden, Dresden, Germany
- Shane Ellis, PhD, Maastricht Multi-Modal Molecular Imaging Institute, Universiteit Maastricht, Maastricht, The Netherlands
- Stephanie Fliedner, PhD, Universitätsklinikum Schleswig-Holstein, Lübeck Medizinische Klinik I, Lübeck, Germany
- Zdenek Fryšák, MD, PhD, University Hospital and Faculty of Medicine and Dentistry, Palacký University Olomouc, Olomouc, Czech Republic
- Hans Ghayee, DO, Department of Internal Medicine, University of Florida, Gainesville, FL
- Garima Gupta, MD, Department of Medicine, Jewish Hospital of Cincinnati, Cincinnati, OH
- Peter Herscovitch, MD, PET Department, Clinical Center, NIH, Bethesda, MD
- W. Marston Linehan, MD, Urologic Oncology Branch, NCI, Bethesda, MD
- Renato Mariani-Constantini, MD, PhD, Ageing Research Center (CeSI), D’Annunzio University, Chieti-Pescara, Italy
- Corina Millo, MD, PET Department, Clinical Center, NIH, Bethesda, MD
- Jirí Neužil, PhD, Institute of Biotechnology, Czech Academy of Sciences, Prague, Czech Republic
- Ondrej Petrak, MD, PhD, Third Department of Medicine, General University Hospital, Prague, Czech Republic
- Margarita Raygada, PhD, Section on Endocrinology and Genetics, NICHD, Bethesda, MD
- Mercedes Robledo, PhD, Human Cancer Genetics Programme, Spanish National Cancer Centre (CNIO), Madrid, Spain
- Douglas Rosing, MD, Translational Medicine Branch, NHLBI, NIH, Bethesda, MD
- Kelly Roszko, MD, PhD, Skeletal Disorders and Mineral Homeostasis Section, NIDCR, Bethesda, MD
- Constantine A. Stratakis, MD, D(med)Sci, Section on Endocrinology and Genetics, NICHD, Bethesda, MD
- Hank Stunnenberg, PhD, Faculty of Science, Radboud Universiteit, Nijmegen, The Netherlands
- Arthur S. Tischler, MD, PhD, New England Medical Center, Boston, MA
- David Torpy, MD, PhD, Royal Adelaide Hospital, Adelaide, Australia
- Richard Tothill, PhD, University of Melbourne Centre for Cancer Research, Melbourne, Australia
- Brad Wood, MD, PhD, Radiology Department, Clinical Center, NIH, Bethesda, MD
- Chunzhang Yang, PhD, Neuro-Oncology Branch, NCI, Bethesda, MD
- Deena Zeltser, MD, Office of the Clinical Director, NICHD, Bethesda, MD
- Zhengping Zhuang, MD, PhD, Neuro-Oncology Branch, NCI, Bethesda, MD
Contact
For more information, email karel@mail.nih.gov or visit http://pheopara.nichd.nih.gov.