Physiological, Biochemical, and Molecular-Genetic Events Governing the Recognition and Resolution of RNA/DNA Hybrids

- Robert J. Crouch, PhD, Head, Section on Formation of RNA
- Susana M. Cerritelli, PhD, Staff Scientist
- Shashikala Mishra, PhD, Postdoctoral Fellow
- Ryo Uehara, PhD, Postdoctoral Fellow
- Kiran Sakhuja, MS, MSc, Biologist
- Yasmeen Ajaj, BS, Postbaccalaureate Fellow
- Emily Helm, BA, BS, Postbaccalaureate Fellow
Damaged DNA is a leading causes of many human diseases and disorders. We study the formation and resolution of RNA/DNA hybrids, which occur during DNA and RNA synthesis. Such hybrid molecules may lead to increased DNA damage but may also play critical roles in normal cellular processes. We are interested in how RNA/DNA hybrids are resolved and in the role that ribonucleases H (RNases H) play in their elimination. Two classes of RNases H, Class I and Class II, are present in most organisms.
Human patients with mutations in RNASEH1 exhibit a typical mitochondrial muscular phenotype [Reference 1]. Our studies were the first to show that RNase H1 is essential for the maintenance of mitochondrial DNA. Mice deleted for the Rnaseh1 gene arrest embryonic development at day 8.5 as a result of failure to amplify mitochondrial DNA [Reference 2]. Aicardi-Goutières syndrome (AGS), a severe neurological disorder with symptoms appearing at or soon after birth, can be caused by defective human RNase H2 [Reference 3]. We are examining mouse models of AGS to gain insight into the human disorder. To understand the mechanisms, functions, substrates, and basic molecular genetics of RNases H, we employ molecular-genetic and biochemical tools in yeast and mouse models.
Contrasts between Class I and Class II RNases H
Many of our investigations over the past few years focused on RNase H1. RNase H1 recognizes the 2′-OH of four consecutive ribonucleotides, while the DNA strand is distorted to fit into a pocket of the enzyme. Thus, the enzyme requires more than one ribonucleotide for cleavage of RNA in RNA/DNA hybrids. In both eukaryotes and prokaryotes, RNases H1 consist of a single polypeptide. In contrast, RNase H2 is a complex of three distinct polypeptides in eukaryotes but a single polypeptide in prokaryotes. The catalytic subunit of the hetero-trimeric RNase H2 of eukaryotes is similar in its primary amino-acid sequence to the prokaryotic enzyme. RNase H2 can recognize and cleave both RNA/DNA hybrids and a single ribonucleotide [Reference 4] or the transition from the ribonucleotide in the case of RNA–primed DNA synthesis (e.g., rrrrrDDDD in DNA—italics indicate transition from ribonucleotide to deoxyribonucleotide).
Several types of RNA/DNA hybrid structures are formed, and they are processed differently. Simple RNA/DNA hybrids consist of one strand of RNA paired with one strand of DNA. The HIV–AIDS reverse transcriptase (RT) forms such hybrids when copying its genomic RNA into DNA. The RT also has an RNase H domain that is structurally and functionally similar to the class I cellular RNase H and is necessary for several steps of viral DNA synthesis. R-loop hybrids (three-stranded nucleic acid structures) have two separated DNA strands, with one hybridized to RNA while the other is in a single-stranded form. Such structures sometimes form during transcription and can lead to chromosomal breakage. However, they are also part of the normal process of switching (recombination) from one form of immunoglobulin to another, resulting in distinct isoforms of antibodies. Another form of hybrid are single or multiple ribonucleotides incorporated into DNA during replication [Reference 4]. The first two types of hybrid are substrates for class I and II RNases H. The third is uniquely recognized by type 2 RNases H.
Dual activities of RNase H2; Aicardi-Goutières syndrome
Eukaryotic RNases H2 recognize and resolve RNA hybridized or covalently attached to DNA—two chemically distinct structures—using the same catalytic mechanism for hydrolysis. RNase H2 mutations that reduce catalytic activity, or fail to properly interact with in vivo substrates, cause Aicardi-Goutières syndrome (AGS). Mutations in seven genes are known to cause AGS, with more than 50% of AGS patients having mutations in any of the three subunits of RNase H2. We previously expressed (in Escherichia coli) and purified human RNases H2 with mutations corresponding to several of those seen in AGS patients; one such mutation, RNASEH2A–G37S (G37S), has significant loss of RNase H2 activity. Using the 3D structure of the human enzyme that we had determined, we could locate all known mutations in RNase H2 that cause AGS. The wide distribution of the mutations suggests that modest changes in stability and in interaction with other unknown proteins, and loss of catalysis can all cause AGS. A mutation near the catalytic center of G37S found in some AGS patients results in low RNase H2 activity for both embedded ribonucleotides in DNA and RNA/DNA hybrids [Reference 3]. We are developing mouse models of AGS to clarify which defects are associated with each RNase H2 activity.
Mice bearing the G37S mutation in homozygous form are perinatal lethal, i.e., either dead at birth or die within a few hours of birth [Reference 3]. Mutations in another gene, TREX1, also cause AGS, and it has been shown that homozygous knockout (KO) mice are viable but die after a few weeks owing to a cardiomyopathy that can be prevented by blocking either an innate or adaptive immune response. In contrast, the G37S–mutant perinatal lethality and the fact that RNase H2 KO mice die during early embryogenesis suggest a more severe defect than that seen in TREX1–KO mice. We attempted to rescue the perinatal phenotype by eliminating one part of the innate immune pathway or by completely inactivating the adaptive immune response. Viability of these mice is no different from that of the innate or adaptive-competent mice. It is possible that there are additional defects in G37S mice that are directly related to viability, not to innate immunity. However, the expression of several interferon-stimulated genes (ISGs) is elevated in mouse embryonic fibroblasts (MEFs) derived from G37S homozygous embryos, supporting a role for innate immunity the AGS phenotype. Damaged DNA that finds its way into the cytoplasm can be sensed by the cGAS protein producing the small molecule cGAMP, which interacts with the Sting protein, an important protein for the DNA–sensing innate immune pathway. Mice that are homozygous for G37S and deleted for the cGAS or Sting genes are mostly perinatal lethal but no longer exhibit increases in ISGs. Interestingly, a small fraction of the double G37S–Sting KO are viable, indicating only limited involvement of ISGs in perinatal lethality [Reference 3]. Further studies are under way, which we expect will lead us to the cause of lethality.
To distinguish the defects that persistent RNA/DNA hybrids and single ribonucleotides joined to DNA cause in vivo, Hyongi Chon, a former postdoctoral fellow, rationally designed a modified RNase H2 to make an enzyme unable to cleave single ribonucleotides embedded in DNA but that retained RNA/DNA hydrolytic activity. The mutant enzyme, which we call RED (Ribonucleotide Excision Deficient), resolves RNA/DNA hybrids, which are substrates of both RNase H1 and RNase H2. Unlike the mouse and human RNases H2, RNase H2 activity is not required in the yeast Saccharomyces cerevisiae. Employing the ease of genetic mutation studies in yeast, we demonstrated that yeast producing the RNase H2 RED enzyme acted in vivo by leaving embedded ribonucleotides (rNMPs) in DNA but was potent in removing RNA in RNA/DNA hybrids.
Embryonic lethality of mice Rnaseh2b–KO stains has been attributed to accumulation of rNMPs in DNA, but lethality could be the result of loss of RNA/DNA hydrolysis or a combination of both rNMP and RNA/DNA hydrolysis defects. To distinguish among the possible causes of embryonic lethality, we generated a mouse that produces the RNase H2RED enzyme. Mouse embryo fibroblasts (MEFs) derived from Rnaseh2RED mice have the same high level of rNMPs as seen in Rnaseh2b–KO MEFs. Interestingly, the Rnaseh2RED mice die around the same time as the Rnaseh2b–KO mice. Therefore, lethality of the Knockout and RED RNase H2 mouse strains results in embryonic death. Rnaseh2aG37S/RED embryos also arrest at approximately the same stage as Rnaseh2aRED/RED embryos because of better association of RNase H2RED than RNase H2G37S with substrate with embedded rNMPs. The result is important because some RNase H2–AGS patients have similar compound heterozygous mutations in which there may be a dominant mutated enzyme.
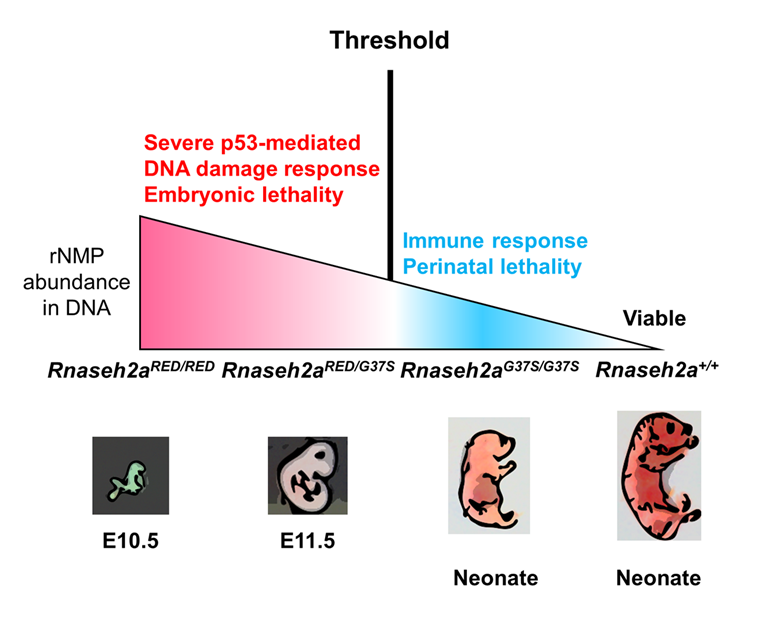
Click image to view.
Figure 1. Two RNase H2 mutants with differential Ribonucleotide Excision Repair activity reveal a threshold of rNMPs for embryonic development.
RNase H2 has two distinct functions: initiation of the Ribonucleotide Excision Repair (RER) pathway by cleaving ribonucleotides (rNMPs) incorporated during DNA replication and processing the RNA portion of an R-loop formed during transcription. An RNase H2 mutant that lacked RER activity but supported R-loop removal revealed that rNMPs in DNA initiate p53–dependent DNA damage response and cause early embryonic arrest in the mouse. However, an RNase H2 AGS–related mutant with residual RER activity develops to birth. Estimations of the number of rNMPs in DNA in these two mutants define a ribonucleotide threshold above which p53 induces apoptosis. Below the threshold, rNMPs in DNA trigger an innate immune response. Cells containing both defective enzymes retain rNMPs above the threshold, indicating competition for RER substrates between active and inactive enzymes and suggesting that patients with compound heterozygous mutations in RNASEH2 genes may not reflect the properties of recombinantly expressed proteins.
Detection of a threshold of ribonucleotide tolerance in DNA for embryonic development
Embryonic development in the absence of RNase H2 exhibits defects as early as E6.5 to 7.5, the period of gastrulation in which cell numbers double every 4–5 h. Previous studies have suggested that the high retention of rNMPs incorporated during DNA replication lead to p53–dependent DNA damage. We provided evidence for the prior speculation that rNMPs are indeed the cause of embryonic lethality. We used mice with a separation of function in the RNase H2 enzyme (RNase H2RED) that retained RNA/DNA hydrolysis but was unable to remove rNMPs in DNA. Embryonic development was arrested at E10, the same day as seen for embryos with complete loss of both of RNase H2’s functions. When there is complete loss of both functions, the abundance of rNMPs in DNA is about 65% of that seen in mouse embryo fibroblast cells. A mouse (RNase H2G37S) with partial loss of both RNase H2 activities develops to birth and retains about 30% of the number of rNMPs in DNA compared with the cells with complete loss of RNase H2. A compound heterozygous mouse in which both RNase H2RED and RNaseh2G37S are present is also early embryonic lethal, retaining about 40% as many rNMPs in as in the deletion cells. Embryos with complete loss of RNase H2, RNase H2RED, and RNase H2RED/G37S all exhibit a p53–dependent DNA–damage response. In contrast, mice with RNase H2G37S develop to birth with little or no p53–dependent DNA damage. The weights of the embryos in which there is p53 DNA damage are only one to a few mg, whereas the RNase HG37S mouse is about 1000 mg at birth, an enormous difference, which indicates more than normal levels of rNMPs do not necessarily cause embryonic lethality. We conclude that a threshold of tolerance of rNMPs in DNA for embryonic development past E10 is exceeded in all mouse strains tested except RNase HG37S. Human patients with the RNASEH2A G37S mutation have Aicardi-Goutières syndrome. Although the patients with RNASEH2A G37S mutations are homozygous, similar to our RNase H2RED/G37S mice, some AGS patients are compound heterozygous, with each allele having a different mutation in the same RNase H2 gene. In vitro studies of mutant forms of RNase H2 mutations reflect the properties of the RNase H2 mutant but may be unreliable for assessing the contribution of each of the two forms of RNase H2 when both are present in vivo. The strong effect on the stage of lethality of RNase H2RED/G37S embryos indicates a competition between the rNMP active RNase H2G37S and the inactive RNase H2RED for some step in removal of rNMPs. The protein proliferating cell nuclear antigen (PCNA) is a critical component in removing rNMPs in DNA. We suggest that the competition between RNase H2RED and RNase H2G37S occurs when RNase H2 interacts with PCNA to repair rNMPs in DNA rather than binding to rNMPs in DNA.
Additional Funding
- NICHD IRP Director's Award
Publications
- Maul RW, Chon H, Sakhuja K, Cerritelli SM, Gugliotti LA, Gearhart PJ, Crouch RJ. R-Loop depletion by over-expressed RNase H1 in mouse B cells increases activation-induced deaminase access to the transcribed strand without altering frequency of isotype switching. J Mol Biol 2017;429:3255-3263.
- Malfatti MC, Balachander S, Antoniali G, Duk Koh K, Saint-Pierre C, Gasparutto D, Chon H, Crouch RJ, Storici F, Tell G. Abasic and oxidized ribonucleotides embedded in DNA are processed by human APE1 and not by RNase H2. Nucleic Acids Res 2017;45:11193–11212.
- Uehara R, Cerritelli SM, Hasin N, Sakhuja K, London M, Iranzo J, Chon H, Grinberg A, Crouch RJ. Two RNase H2 mutants with differential rNMP processing activity reveal a threshold of ribonucleotide tolerance in DNA for embryonic development. Cell Rep 2018;25:1135-1145.
Collaborators
- Frederic Chedin, PhD, University of California Davis, Davis, CA
- Patricia J. Gearhart, PhD, Laboratory of Molecular Biology and Immunology, NIA, Baltimore, MD
- Ian J. Holt, PhD, Biodonostia Institute, Donostia, San Sebastián, Spain
- Herbert C. Morse, MD, Laboratory of Immunopathology, NIAID, Bethesda, MD
- Francesca Storici, PhD, School of Biological Sciences, Georgia Institute of Technology, Atlanta, GA
- Nan Yan, PhD, University of Texas Southwestern Medical Center, Dallas, TX
- Kiyoshi Yasukawa, PhD, Kyoto University, Kyoto, Japan
Contact
For more information, email crouch@helix.nih.gov or visit http://sfr.nichd.nih.gov.