Protein Sorting in the Endomembrane System

- Juan S. Bonifacino, PhD, Head, Section on Intracellular Protein Trafficking
- Rafael Mattera, PhD, Staff Scientist
- Xiaolin Zhu, RN, Technician
- Jing Pu, PhD, Research Fellow
- Raffaella De Pace, PhD, Visiting Fellow
- David Gershlick, PhD, Visiting Fellow
- Carlos M. Guardia, PhD, Visiting Fellow
- Morie Ishida, PhD, Visiting Fellow
- Rui Jia, PhD, Visiting Fellow
- Tal Keren-Kaplan, PhD, Visiting Fellow
- Elodie Mailler, PhD, Visiting Fellow
- Amra Saric, PhD, Visiting Fellow
- Arianne Foster, BS, Postbaccalaureate Student
- Akansha Jain, BS, Postbaccalaureate Student
Our lab investigates the molecular mechanisms by which transmembrane proteins (referred to as “cargo”) are sorted to different compartments of the endomembrane system in eukaryotic cells. In polarized cells, such as epithelial cells and neurons, the system comprises an array of membrane-enclosed organelles including the endoplasmic reticulum (ER), the Golgi apparatus, the trans-Golgi network (TGN), endosomes, lysosomes, lysosome-related organelles (LROs) (e.g., melanosomes), and various domains of the plasma membrane. Transport of cargo between these compartments is mediated by carrier vesicles or tubules that bud from a donor compartment, translocate through the cytoplasm, and eventually fuse with an acceptor compartment. Work in our laboratory focuses on the molecular machineries that mediate these processes, including: (1) sorting signals and adaptor proteins that select cargo proteins for packaging into the transport carriers; (2) microtubule motors and organelle adaptors that drive movement of the transport carriers and other organelles through the cytoplasm; and (3) tethering factors that promote fusion of the transport carriers to acceptor compartments. We study the machineries in the context of various intracellular transport pathways, including endocytosis, recycling to the plasma membrane, retrograde transport from endosomes to the TGN, biogenesis of lysosomes and LROs, and polarized sorting in epithelial cells and neurons. We apply knowledge gained from this basic research to the elucidation of disease mechanisms, including congenital disorders of protein traffic, such as the pigmentation and bleeding disorder Hermansky-Pudlak syndrome (HPS) and hereditary spastic paraplegias (HSPs), and to the exploitation of intracellular transport by pathogens such as HIV-1.
AP-4 mediates export of ATG9A from the TGN to promote autophagosome formation.
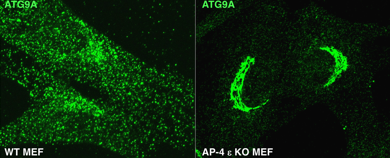
This past year, we discovered a role for the heterotetrameric AP-4 (adaptor protein 4) complex in the signal-mediated export of the autophagy protein ATG9A from the TGN, which contributes to the elucidation of the pathogenesis of a group of HSPs caused by mutations in AP-4 subunit genes. The HSPs are a clinically and genetically heterogeneous group of disorders characterized by progressive lower limb spasticity. Mutations in any of the four subunits of AP-4 cause an autosomal recessive form of complicated HSP referred to as "AP-4 deficiency syndrome." In addition to lower limb spasticity, the syndrome features intellectual disability, microcephaly, seizures, a thin corpus callosum, and upper limb spasticity. To elucidate the pathogenetic mechanism, we characterized a knockout (KO) mouse for the AP4E1 gene encoding the epsilon subunit of AP-4. We found that AP-4 epsilon KO mice exhibit a range of neurological abnormalities, including hindlimb clasping, reduced motor coordination, and weak grip strength. In addition, the KO mice display a thin corpus callosum and axonal swellings in various areas of the brain and spinal cord. Biochemical and cellular analyses identified ATG9A, the only multispanning membrane component of the core autophagy machinery, as a specific AP-4 cargo. Moreover, we found that AP-4 promotes signal-mediated export of ATG9A from the TGN to the peripheral cytoplasm, contributing to lipidation of the autophagy protein LC3B and maturation of pre-autophagosomal structures. Immunohistochemical analyses of various AP-4–deficient cell types, including non-neuronal cell lines, patient skin fibroblasts, and mouse neurons, showed retention of ATG9A at the TGN and its depletion from the peripheral cytoplasm (Figure 1). ATG9A mislocalization was associated with an increased tendency to accumulate mutant huntingtin (HTT) aggregates in the axons of AP-4 epsilon KO neurons. The findings indicated that the AP-4 epsilon KO mouse is a suitable animal model for human AP-4 deficiency syndrome and that defective mobilization of ATG9A from the TGN and impaired autophagic degradation of protein aggregates might contribute to neuroaxonal dystrophy in this disorder.
Click image to view.
Figure 1. AP-4 epsilon KO causes retention of ATG9A at the TGN of mouse embryonic fibroblasts.
Segregation in the Golgi complex precedes export of endolysosomal proteins in distinct transport carriers, independently of AP complexes.
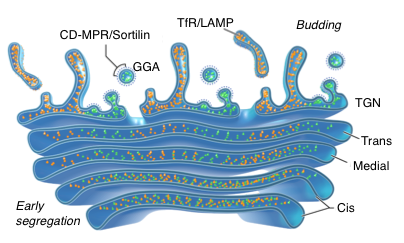
The studies described above demonstrated that ATG9A requires AP-4 for its signal-dependent export from the TGN. It has been proposed that other biosynthetic cargos destined for the endolysosomal system exit the TGN by interaction of sorting signals with adaptor proteins. However, there was no direct evidence for such a mechanism. Using advanced imaging methodologies, we found that this is indeed the case for the cation-dependent mannose 6-phoshate receptor (CD-MPR) and sortilin, which require interaction with the GGA adaptor proteins for export from the TGN into transport carriers bound for the endosomal system. In contrast, we found that the transferrin receptor (TfR) and the lysosomal protein LAMP1 exit the TGN independently of sorting signals and adaptor proteins. Moreover, they were transported into a different type of carrier directed towards the plasma membrane rather than to endosomes and lysosomes. These proteins subsequently undergo AP-2–dependent endocytic delivery to their corresponding compartments. Strikingly, we observed that these different TGN export mechanisms are preceded by early segregation of the corresponding proteins within distinct domains of the Golgi stack by virtue of the luminal and transmembrane domains of the proteins (Figure 2). The findings revealed a diversity of sorting mechanisms in the Golgi complex, including early segregation in the Golgi stack prior to export into distinct populations of transport carriers.
Click image to view.
Figure 2. TfR/LAMP are segregated from CD-MPR/Sortilin in the Golgi stack before their budding by AP–independent and –dependent mechanisms, respectively.
Function of the BORC complex in the regulation of lysosome movement
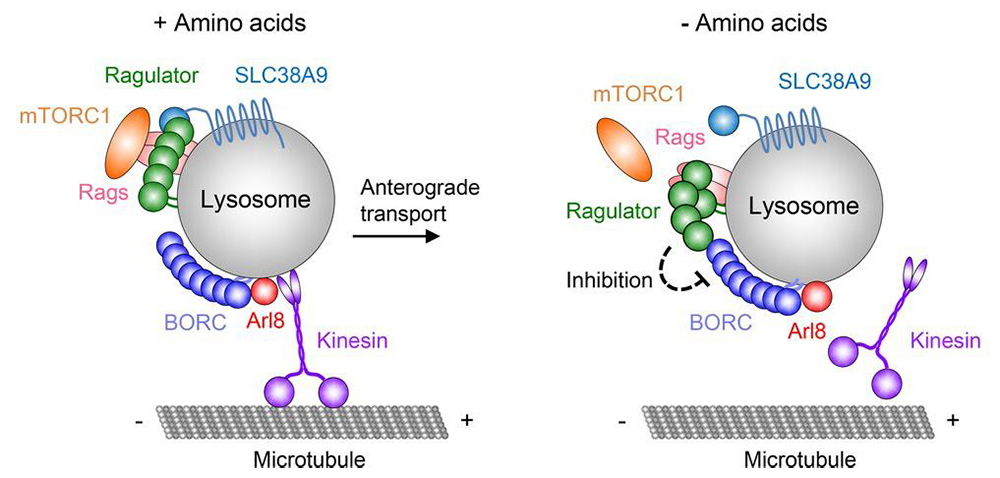
Another important accomplishment of the lab was the discovery of the BORC complex and its role in lysosome positioning. The multiple functions of lysosomes are critically dependent on their ability to move bidirectionally along microtubules between the center and the periphery of the cell. Centrifugal and centripetal movement of lysosomes are mediated by kinesin and dynein motors, respectively. We recently discovered a multi-subunit complex named BORC, which recruits the small GTPase ARL8 to lysosomes to promote their kinesin-dependent movement towards the cell periphery (Figure 3). We showed that BORC and ARL8 function upstream of two structurally distinct kinesin types: kinesin-1 (KIF5B) and kinesin-3 (KIF1B and KIF1A). Remarkably, KIF5B and KIF1B/KIF1A move lysosomes along different microtubule tracks. The findings establish BORC as a master regulator of lysosome positioning through coupling to different kinesins and microtubule tracks.
Click image to view.
Figure 3. Hypothetical model for the regulation of lysosome positioning by Ragulator and BORC.
Interaction of BORC with Ragulator controls lysosome positioning in response to amino acid availability.
This past year, we discovered an additional role for BORC in the response of lysosomes to amino-acid deprivation. Depletion of amino acids from the medium turns off a signaling pathway involving the Ragulator complex (a pentameric protein complex that is required for mTORC1 activation) and the Rag GTPases, causing inactivation of the mammalian target of rapamycin complex 1 (mTORC1, a nutrient/energy/redox sensor that controls protein synthesis) kinase from the lysosomal membrane. Reduced phosphorylation of mTORC1 substrates inhibits protein synthesis while activating autophagy. Amino-acid depletion also causes clustering of lysosomes in the juxtanuclear area of the cell, but the mechanisms responsible for this phenomenon were poorly understood. We found that Ragulator directly interacts with BORC, inhibiting its ability to drive lysosomes towards the cell periphery. Amino-acid depletion strengthens this interaction, explaining the redistribution of lysosomes to the juxtanuclear area. The findings demonstrated that amino-acid availability controls lysosome positioning through Ragulator-dependent modulation of BORC (Figure 3).
Hijacking of the retromer complex by Legionella pneumophila
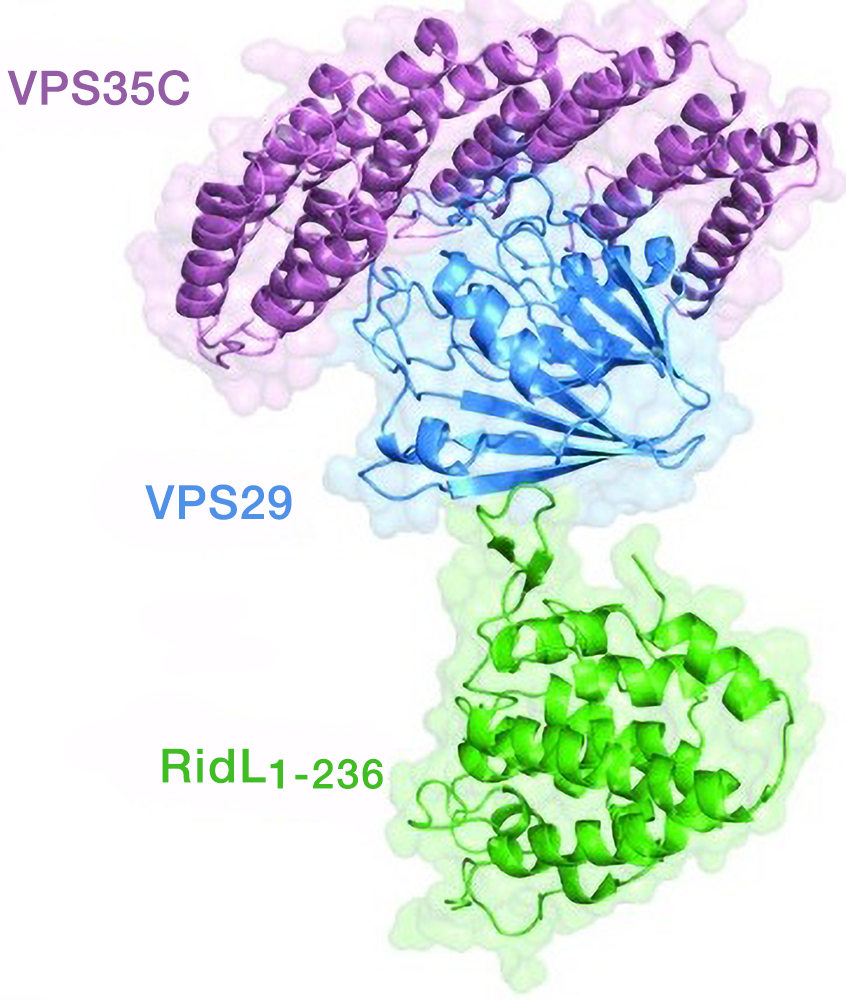
We collaborated with Aitor Hierro’s group to characterize the mechanism by which the intracellular pathogen Legionella pneumophila hijacks retromer, a complex composed of VPS26, VPS29, and VPS35 subunits and involved in protein recycling from endosomes. The hijacking involves a L. pneumophila effector named RidL, which interacts with the VPS29 subunit of retromer. We solved the crystal structure of RidL in complex with a VPS29-VPS35 retromer subcomplex (Figure 4). We found that a hairpin loop protruding from RidL inserts into a conserved pocket on VPS29 that is also used by cellular ligands, such as TBC1D5 and VARP, for VPS29 binding. Consistent with the idea of molecular mimicry in protein interactions, RidL outcompeted TBC1D5 for binding to VPS29. Furthermore, we found that the interaction of RidL with retromer does not interfere with retromer dimerization but is essential for association of RidL with retromer-coated vacuolar and tubular endosomes. The work thus provided structural and mechanistic evidence for how RidL is targeted to endosomal membranes.
Click image to view.
Figure 4. Structure of RidL in complex with retromer.
Additional Funding
- Intramural AIDS Targeted Antiviral Program (IATAP)
Publications
- De Pace R, Skirzewski M, Damme M, Mattera R, Mercurio J, Foster AM, Cuitino L, Jarnik M, Hoffmann V, Morris HD, Han TU, Mancini GMS, Buonanno A, Bonifacino JS. Altered distribution of ATG9A and accumulation of axonal aggregates in neurons from a mouse model of AP-4 deficiency syndrome. PLoS Genet 2018;14:e1007363.
- Guardia CM, De Pace R, Mattera R, Bonifacino JS. Neuronal functions of adaptor complexes involved in protein sorting. Curr Opin Neurobiol 2018;51:103-110.
- Mattera R, Park SY, De Pace R, Guardia CM, Bonifacino JS. AP-4 mediates export of ATG9A from the trans-Golgi network to promote autophagosome formation. Proc Natl Acad Sci USA 2017;114:E10697-E10706.
- Pu J, Keren-Kaplan T, Bonifacino JS. A Ragulator-BORC interaction controls lysosome positioning in response to amino acid availability. J Cell Biol 2016;216:4183-419.
- Chen Y, Gershlick DC, Park SY, Bonifacino JS. Segregation in the Golgi complex precedes export of endolysosomal proteins in distinct transport carriers. J Cell Biol 2017;216:4141-4151.
Collaborators
- Aitor Hierro, PhD, CIC bioGune, Derio, Spain
Contact
For more information, email bonifacinoj@helix.nih.gov or visit https://irp.nih.gov/pi/juan-bonifacino.