Interplay between Membrane Organelles, Cytoskeleton, and Metabolism in Cell Organization and Function

- Jennifer Lippincott-Schwartz, PhD, Head, Advanced Microscopy Core
- Chad D. Williamson, PhD, Research Fellow
- Dani Cai, PhD, Visiting Fellow
- Dvir Gur, PhD, Visiting Fellow
- Yang Niu, PhD, Visiting Fellow
- Lingfeng Chen, PhD, Volunteer
We investigate the global principles underlying cell behavior at both small and large spatial scales. At the small scale, we employ the super-resolution imaging techniques of photoactivated localization microscopy (PALM), interferometric 3D PALM, single-particle tracking PALM, and pair-correlation PALM to map the spatial organization, stoichiometry, and dynamics of proteins associated with various membrane-bound compartments and with the cytoskeleton. We also employ fluorescence photobleaching, photoactivation, fluorescence correlation, and fluorescence energy transfer methods to measure protein-protein interactions, protein turnover rates, and protein association rates. Such approaches allow us to assay cellular functions, including receptor stoichiometry and protein clustering and diffusion behavior at the nanometric scale in living cells. At the large scale, we investigate how complex behaviors of cells arise, such as cell crawling, polarization, cytokinesis, and viral budding. We study such complex behaviors by quantitatively analyzing diverse intracellular processes, including membrane trafficking, autophagy, actin/microtubule dynamics, and organelle assembly/disassembly pathways, which undergo dramatic changes as cells alter their behavior and organization throughout life. To support these efforts, we combine various fluorescence-based imaging approaches, including total internal reflection fluorescence (TIRF) microscopy imaging and spinning-disk and laser-scanning confocal microscopy, with FRAP (fluorescence recovery after photobleaching), FLIP (fluorescence loss in photobleaching), and photoactivation to obtain large image data sets. We process the data sets computationally to extract biochemical and biophysical parameters, which can be related to the results of conventional biochemical assays. We then use the results to generate mechanistic understanding and predictive models of the behavior of cells and subcellular structures (including endoplasmic reticulum, Golgi, cilia, endosomes, lysosomes, autophagosomes, and mitochondria) under healthy and pathological conditions.
Click image to view.
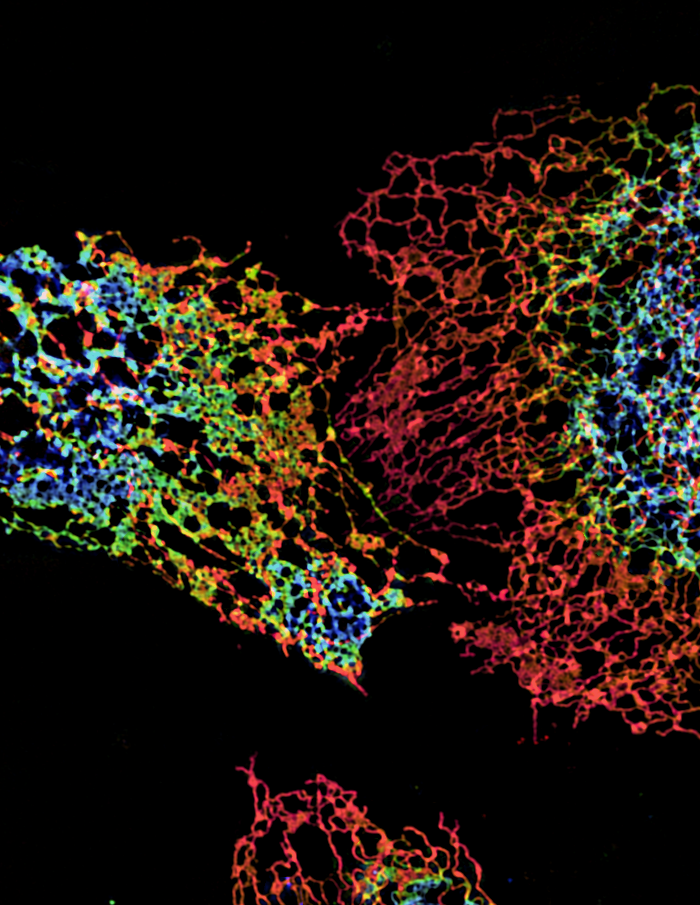
ER structure visualized using structured illumination microscopy (SIM)
Structure of peripheral endoplasmic reticulum (ER) in three cells labeled with an ER protein marker visualized by three-dimensional SIM. The color coding represents different z heights of the ER.
Spectral unmixing for simultaneous analysis of multiple fluorophores within cells
The ability to unambiguously distinguish more than a few different labels in a single fluorescence image has been severely hampered by the excitation cross-talk and emission bleed-through of fluorophores with highly overlapping spectra. To overcome the problem, we developed a cell-labeling, image-acquisition, and image-analysis approach to study the spatial distribution of six different organelles within eukaryotic cells. The work allowed us to present a systems-level analysis of the organelle interactome using a multispectral image-acquisition method that overcomes the challenge of spectral overlap in the fluorescent protein palette. We employed confocal and lattice light sheet (LLS) instrumentation and an imaging informatics pipeline of five steps to achieve mapping of organelle numbers, volumes, speeds, positions, and dynamic inter-organelle contacts in live fibroblast cells. Cells were transfected with six fluorescent fusion protein markers of organelles or labeled with compartment-specific fluorescent chemical dyes to highlight six subcellular compartments: peroxisomes, lysosomes, ER, mitochondria, Golgi, and lipid droplets (LD). We acquired live-cell, time-lapse images and then applied linear unmixing algorithms to every pixel in the image, deconvolving spectrally-overlapping fluorophores. Our data allowed us to describe the frequency and locality of two-, three-, four-, and five-way interactions among six different membrane-bound organelles (ER, Golgi, lysosome, peroxisome, mitochondria, and LD) and to show how these relationships change over time. We further demonstrated that each organelle has a characteristic distribution and dispersion pattern in three-dimensional space and that there is a reproducible pattern of contacts among the six organelles, impacted by microtubule and cell nutrient status. In this way, we provided a full systems-level description of the spatial organization of eukaryotic organelles under various physiological conditions. The live-cell confocal and LLS spectral-imaging approaches we developed in this study are applicable to any cell system expressing multiple fluorescent probes, whether in normal conditions or when cells are exposed to disturbances such as drugs, pathogens, or stress. The methodology thus offers a powerful new descriptive tool and source for hypotheses testing in the fields of cellular organization and dynamics.
ER exit sites regulate procollagen quality control through non-canonical autophagy.
Type I collagen is the main component of bone matrix and other connective tissues. Rerouting of its procollagen precursor to a degradative pathway is crucial for osteoblast survival in pathologies involving excessive intracellular buildup of procollagen that is improperly folded and/or trafficked. What cellular mechanisms underlie this rerouting had been unclear. We studied these mechanisms in collaboration with the lab of Sergey Leikin, employing live-cell imaging and correlative light and electron microscopy (CLEM) to examine procollagen trafficking both in wild-type mouse osteoblasts and osteoblasts expressing a bone pathology–causing mutant procollagen. We found that, although most procollagen molecules successfully trafficked through the secretory pathway in these cells, a subpopulation did not. The latter molecules appeared in numerous dispersed puncta colocalizing with subunits of the COPII coatomer, autophagy markers, and ubiquitin machinery, with more puncta seen in mutant procollagen-expressing cells. Blocking endoplasmic reticulum exit site (ERES) formation suppressed the number of these puncta, suggesting they formed after procollagen entry into ERESs. The punctate structures containing procollagen, COPII, and autophagic markers did not move toward the Golgi but instead were relatively immobile. They appeared to be quickly engulfed by nearby lysosomes through a bafilomycin-insensitive pathway. CLEM and fluorescence recovery after photobleaching experiments suggested that engulfment occurred through a noncanonical form of autophagy resembling microautophagy of ERESs. The findings point to a non-canonical mode of procollagen quality control at ERESs involving lysosomal engulfment of ERESs that have been modified with autophagic machinery.
ER structure and dynamics visualized with increased spatio-temporal resolution
The endoplasmic reticulum (ER) consists of interconnected tubules and flattened sheets that extend from the nuclear envelope to the periphery of the cell, impacting every cellular compartment through its contacts and functional interactions. Mutations in proteins that regulate the shape of the ER lead to various neurological disorders. We employed five different super-resolution technologies with complementary strengths and weaknesses in spatial and temporal capabilities to study the fine morphology and dynamics of the peripheral ER. A high-speed variation of structured illumination microscopy (SIM) allowed ER dynamics to be visualized at unprecedented speeds and resolution. Three-dimensional SIM (3D-SIM) and Airyscan imaging allowed comparison of the fine distributions of different ER–shaping proteins. Lattice light sheet point accumulation for imaging in nanoscale topography (LLS-PAINT) and focused ion-beam scanning electron microscopy (FIB-SEM) permitted 3D characterization of various ER structures. Using these approaches, we observed that many ER structures previously thought to be flat membrane sheets are instead densely packed tubular arrays, which we call ER matrices. The matrices were extremely compact, with spaces between tubules below the resolving power of most super-resolution methodologies. We also discovered that ER tubules and junctions undergo fast oscillations, rapidly interconverting from tight to loose arrays. The oscillations of tubules and junctions were energy-dependent and allowed the ER to interconvert between tight and loose tubule networks. Our discovery of dense tubular matrices in areas previously thought of as flat sheets provides a new model for maintaining and generating ER structure. In this model, ER matrices would sequester excess membrane proteins and lipids, and their dynamic interconversion into loose tubule arrays would permit the ER to rapidly extend its shape to reach the cell periphery, for example during cell locomotion.
Publications
- Omari S, Makareea E, Roberts-Pilgrim A, Mirigian L, Jarnik M, Ott C, Lippincott-Schwartz J, Leikin S. Noncanonical autophagy at ER exit sites regulates procollagen turnover. Proc Natl Acad Sci USA 2018;115:E10099–E10108.
- Cohen S, Rambold AS, Lippincott-Schwartz J. Mitochondrial and lipid droplet dynamics regulate intra- and intercellular fatty acid trafficking. Mol Cell Oncol 2018;5:e1043038.
- Cai D, Sukenik S, Feliciano D, Gruebele M, Lippincott-Schwartz J. Phase separation of YAP reprograms cells for long-term YAP target gene expression. bioRxiv 2018;438416.
- Valm AM, Cohen S, Legant WR, Melunis J, Hershberg U, Wait E, Cohen AR, Davidson MW, Betzig E, Lippincott-Schwartz J. Applying systems-level spectral imaging and analysis to reveal the organelle interactome. Nature 2017;546(7656):162-167.
- Ritter AT, Kapnick SM, Murugesan S, Schwartzberg PL, Griffiths GM, Lippincott-Schwartz J. Cortical actin recovery at the immunological synapse leads to termination of lytic granule secretion in cytotoxic T lymphocytes. Proc Natl Acad Sci USA 2017;114(32):E6585-E6594.
Collaborators
- Eric Betzig, PhD, Howard Hughes Medical Institute, Janelia Farm Research Campus, Ashburn, VA
- Craig Blackstone, MD, PhD, Cell Biology Section, NINDS, Bethesda, MD
- Harald Hess, PhD, Howard Hughes Medical Institute, Janelia Farm Research Campus, Ashburn, VA
- Sergey Leikin, PhD, Section on Physical Biochemistry, NICHD, Bethesda, MD
- Uri Manor, PhD, Salk Institute for Biological Studies, La Jolla, CA
- Mark P. Matteson, PhD, Laboratory of Neurosciences, NIA, Baltimore, MD
- Jonathon J. Nixon-Abell, PhD, Cell Biology Section, NINDS, Bethesda, MD
- George Patterson, PhD, Section on Biophotonics, NIBIB, Bethesda, MD
- Alex T. Ritter, PhD, Genentech, San Francisco, CA
- Bennett Waxse, MD, PhD, University of Chicago Medicine, Chicago, IL
- Pamela J. Yao, PhD, Cellular and Molecular Neurosciences Section, NIA, Baltimore, MD
Contact
For more information, email lippincj@mail.nih.gov or visit http://lippincottschwartzlab.nichd.nih.gov.