
Exploring Genes and Signals Regulating Mammalian Hematopoiesis and New Approaches to Immunotherapy

- Paul E. Love,
MD, PhD, Head, Section on Hematopoiesis and Lymphocyte Biology - LiQi Li, MD, PhD, Staff Scientist
- Guillaume Gaud, PhD, Research Fellow
- Seeyoung Choi, PhD, Senior Fellow
- Avik Dutta, PhD, Visiting Fellow
- Teri Hatzihristidis, PhD, Visiting Fellow
- Sitanshu Sarangi, PhD, Visiting Fellow
- Dalal El-Khoury, BS, Technician
- Jan Lee, BS, Technician
- Jayna Bryant, BS, Postbaccalaureate Fellow
Our research focuses primarily on the development of the mammalian hematopoietic system. A long-term area of interest for our laboratory is the study of signal-transduction molecules and pathways that regulate T lymphocyte (T cell) maturation in the thymus. A critical signaling protein complex called the T cell antigen receptor (TCR) is responsible for T cell antigen recognition and is also necessary for T cell development. Currently, we are analyzing transgenic and conditional non-signaling mutants of the TCR signaling subunits, generated in our lab, to evaluate the importance of individual TCR signaling chains and signaling motifs at specific stages of T cell development and in mature T cells. In current studies, we are investigating whether modification of TCR signaling subunits can be used to enhance the tumoricidal activity of T cells for cancer treatment. Using gene profiling, we seek to identify proteins that are important for "fine-tuning" the T cell signaling response in developing and mature T lymphocytes. In conjunction with checkpoint inhibitors for immunotherapy in humans, such molecules may also be targets to enhance T cell anti-tumor activity. We also investigate the function of novel T cell–specific proteins that we identified by subtraction-library screening or RNA-Seq. Our studies revealed a critical role for one of these proteins, called THEMIS, in T cell development by enhancing the TCR–signaling response in thymocytes. We are currently investigating the function of other THEMIS family members, THEMIS2 and THEMIS3, which are expressed in B and myeloid cells or in intestine epithelial cells, respectively. We found that another newly identified protein expressed in maturing T cells, Fbxl12, is important for regulating proliferation during early T cell development by coordinating pre–TCR and Notch signaling.
Another long-term area of investigation focuses on a transcription complex that regulates hematopoietic stem cell (HSC) maintenance and early stages of T cell, B cell, and erythrocyte development. We have begun characterizing a protein (Ldb1) that is important for the generation and maintenance/self-renewal of HSCs, which revealed a critical function for Ldb1 as a key subunit of multimeric DNA–binding complexes in controlling the self-renewal/differentiation cell-fate decision in HSCs. Current work, which involves genome-wide screening by single cell RNA-Seq and ChIP-Seq, seeks to extend our knowledge of the role of Ldb1 complexes in regulating gene transcription and to explore the function of such complexes in other hematopoietic lineages. We have also begun to investigate the importance of Ldb1 complexes in regulating self-renewal in immature thymocytes and in the induction of T cell acute lymphoblastic leukemia (T-ALL). Our results suggest that Ldb1 complexes represent potential therapeutic targets for the treatment of an aggressive form of childhood leukemia called early T progenitor (ETP) acute lymphoblastic leukemia (ETP-T-ALL).
T cell antigen receptor (TCR) signaling in thymocyte development
Much of our research in the past has focused on the role of TCR signal transduction in thymocyte development. Signal transduction sequences, termed immuno-receptor tyrosine-based activation motifs (ITAMs), are contained within four distinct subunits of the multimeric TCR complex (CD3-zeta, CD3-gamma, CD3-delta, and CD3-epsilon). Di-tyrosine residues within ITAMs are phosphorylated upon TCR engagement; their function is to recruit signaling molecules, such as protein tyrosine kinases, to the TCR complex, thereby initiating the T cell–activation cascade. Although conserved, ITAM sequences are non-identical, raising the possibility that the diverse developmental and functional responses controlled by the TCR may be partly regulated by distinct ITAMs through the recruitment of different effector molecules. This question has been studied for years by many laboratories but, to date, a clear functional characterization of ITAM motifs in TCR signaling and T cell effector functions has not been established. We previously generated CD3-zeta–deficient and CD3-epsilon–deficient mice by gene targeting. We genetically reconstituted these mice with transgenes encoding wild-type or signaling-deficient (ITAM–deleted) forms of CD3-zeta and CD3-epsilon and characterized the developmental and functional consequences of the alterations in TCR signaling potential. We found that TCR ITAMs appear to be functionally equivalent, but act in concert to amplify TCR signals, and that TCR signal amplification is important for thymocyte selection, the process by which potentially useful immature T cells are instructed to survive and differentiate further (positive selection) and by which potentially auto-reactive cells, which may cause autoimmune disease, are eliminated in the thymus (negative selection).
Figure 1. T cell antigen receptors expressed in 6Y/6Y and 6F/6F knock-in mice
Subunit composition of the T cell antigen receptors in 6Y/6Y and 6F/6F mice. 6Y/6Y mice express wild-type zeta chain dimers with functional ITAM signaling motifs that contain two tyrosine (Y) residues. 6F/6F mice express mutant zeta chain dimers in which the ITAM tyrosines have been changed to phenylalanine (F).
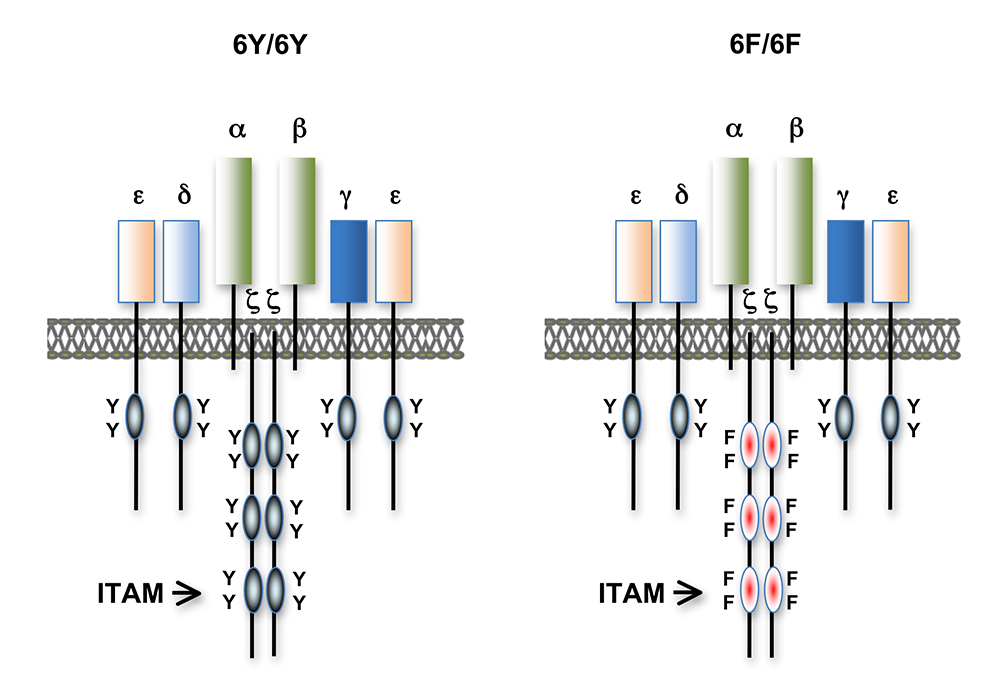
Figure 1. T cell antigen receptors expressed in 6Y/6Y and 6F/6F knock-in mice
Subunit composition of the T cell antigen receptors in 6Y/6Y and 6F/6F mice. 6Y/6Y mice express wild-type zeta chain dimers with functional ITAM signaling motifs that contain two tyrosine (Y) residues. 6F/6F mice express mutant zeta chain dimers in which the ITAM tyrosines have been changed to phenylalanine (F).
We also found that a complete complement of TCR ITAMs is not required for most mature T cell effector functions. However, our recent work demonstrated a requirement for ITAM multiplicity for the generation of T follicular helper cells, which are necessary for optimal B cell antibody responses. One possible explanation for the relatively mild phenotype observed in the TCR ITAM–reduced mice is that ITAM–mediated signal amplification is not required for most mature T cell activation responses; another is that some ITAMs perform more subtle, regulatory functions. To investigate this question in more detail, we recently generated a TCR zeta chain conditional 'knock-in' mouse in which T cell development and selection can occur without attenuation of TCR signaling (i.e., in the presence of a wild-type 3-ITAM ‘6Y’ zeta chain), but in which mature, post-selection T cells may be induced to express TCRs containing signaling-defective (0-ITAM ‘6F’) zeta chains [where the six critical ITAM tyrosines (Y) are mutated to phenylalanine (F)] in lieu of wild-type (6Y) zeta chains (Figure 1). In this experimental model, TCR signaling in mature T cells should not be influenced by potential regulatory mechanisms that operate during T cell maturation to compensate for the reduced signaling potential of 6F TCRs; thus, T cells in such mice should be faithful indicators of the role of multiple TCR ITAMs in mediating specific, mature T cell responses. We confirmed that the knock-in zeta locus functions as predicted. We next evaluated the effect of late ‘switching’ from 6Y zeta to non-signaling 6F zeta by Cre recombinase expression in mature T cells generated with wild-type 6Y zeta–containing TCRs and found that the phenotype was identical to germline inactivation of zeta ITAMs, demonstrating that compensation does not explain the mild phenotype of zeta 6F mice. Unexpectedly, we discovered an inhibitory role for zeta ITAM signaling in mature T cell responses to weak (low-affinity) antigens that was attributable to the recruitment of the inhibitory tyrosine phosphatase SHP-1 by mono-phosphorylated zeta ITAMs. Strikingly, inactivation of the zeta ITAMs (switching from 6Y to 6F zeta) resulted in enhanced TCR signaling and enhanced T cell effector functions when the TCR was engaged by low-affinity ligands, but functional 6Y zeta ITAMs contributed positively to signaling by high-affinity ligands. This revealed a dual (activating and inhibitory) context–dependent function for zeta ITAMs in TCR signaling depending on the affinity of the TCR–ligand interaction. Given that most tumor-specific antigens are low affinity and that this property limits current TCR–based approaches to tumor immunotherapy, we explored the effect of zeta ITAM inactivation on T cell–tumoricidal activity. Notably, we found that inactivation of zeta ITAMs markedly enhanced T cell–tumoricidal activity against low-affinity tumor antigens and improved tumor killing (Figure 2), results that provide information relevant to the optimal design of engineered tumor antigen–specific TCRs and, possibly, chimeric antigen receptors (CARs), which are currently configured to express only the wild-type 6Y zeta signaling molecule. Our results call for a revision of the current paradigm of TCR signaling, which assumes that all ITAMs perform only activating roles in TCR signaling, and identify a novel counter-intuitive approach to T cell–based tumor immunotherapy.
Figure 2. Expression of TCRs that include non-signaling (6F) CD3zeta subunits unexpectedly results in enhanced CD8 T cell–mediated tumor killing in response to low affinity antigens.
T/B cell deficient Rag2–/– mice were injected with OTI (ovalbumin peptide–specific) TCR transgenic CD8+ T cells that express 6Y (OTI-6Y) or 6F (OTI-6F) CD3zeta TCR subunits 7 days after B16F10-V4 melanoma implantation. The OTI TCR binds with low affinity to V4 peptide expressed by B16F10-V4 melanoma cells (V4 peptide acts as a tumor-specific antigen in these experiments). Left. Measurement of tumor size over time. Right. Survival plots of experiment shown on left. N=5 biological replicates. Results shown are representative of 3 experiments. Data are represented as mean ± SD. **P < 0.01, ****P < 0.001.
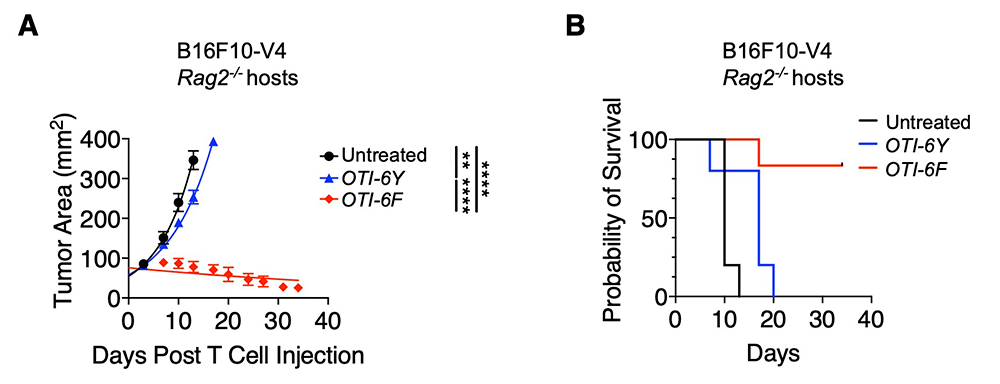
Figure 2. Expression of TCRs that include non-signaling (6F) CD3zeta subunits unexpectedly results in enhanced CD8 T cell–mediated tumor killing in response to low affinity antigens.
T/B cell deficient Rag2–/– mice were injected with OTI (ovalbumin peptide–specific) TCR transgenic CD8+ T cells that express 6Y (OTI-6Y) or 6F (OTI-6F) CD3zeta TCR subunits 7 days after B16F10-V4 melanoma implantation. The OTI TCR binds with low affinity to V4 peptide expressed by B16F10-V4 melanoma cells (V4 peptide acts as a tumor-specific antigen in these experiments). Left. Measurement of tumor size over time. Right. Survival plots of experiment shown on left. N=5 biological replicates. Results shown are representative of 3 experiments. Data are represented as mean ± SD. **P < 0.01, ****P < 0.001.
Identification and characterization of TCR–tuning proteins that may serve as targets for immunotherapy
We extended our analysis of TCR signaling subunits to other molecules that may participate in or influence the TCR signaling response. The cell-surface protein CD5 negatively regulates TCR signaling, and functions in thymocyte selection and in mature T cell responses. Examination of CD5 expression during T cell development revealed that surface levels of CD5 are regulated by TCR signal intensity and by the affinity of the TCR for self-peptide ligands in the thymus that mediate selection. To determine whether the ability to regulate CD5 expression is important for thymocyte selection, we generated transgenic mice that constitutively express high levels of CD5 throughout development. Over-expression of CD5 significantly impaired positive selection of some thymocytes (those that would normally express low levels of CD5) but not of others (those that would normally express high levels of CD5). The findings support a role for CD5 in modulating TCR signal transduction, thereby influencing the outcome of thymocyte selection, a function we termed "TCR tuning." Current studies center on identifying the mechanism by which CD5 inhibits TCR signaling and on determining whether the protein's regulated expression during development is important for preventing autoimmunity. For that purpose, we generated a conditional CD5–deletion mouse in which CD5 expression can be removed before, during, or after T cell development. The ability of individual thymocytes to regulate CD5 expression represents a mechanism for ‘fine tuning’ the TCR–signaling response during development, so that the integrated signaling response can be adjusted to permit T cell functional competency without causing autoimmunity. Reasoning that, in addition to CD5, other molecules participate in TCR tuning, we initiated microarray-based screening for genes differentially expressed in developing T cells under conditions of high- or low-affinity TCR interactions. We identified several genes from this screen for further study, including the gene encoding CD6, a surface receptor that is structurally similar to CD5. We are currently validating the function of these and other putative tuning molecules. Given that ‘tuning’ molecules regulate TCR signaling, they represent potential autoimmune disease–susceptibility markers and potential targets for treatment of patients with cancer, similar to current ‘checkpoint inhibitor’ therapies, which are based on blocking the function of the induced inhibitory molecules PD-1 and CTLA-4. Experiments are under way to investigate this translational potential.
Identification and characterization of Themis family proteins
Using genetic screening, we identified Themis, now known as Themis1, a novel T cell–specific adapter protein (Figure 3). To investigate the function of Themis1 in T cell signaling and development, we generated Themis1 knock-down cell lines, Themis1 knock-out mice (conventional and conditional), and Themis1 transgenic mice. Analysis of the effects of modulating Themis1 expression revealed a critical role for the protein in positive selection and late T cell development. In a collaboration with Richard Cornall, we also investigated the phenotype of Themis2 knockout mice generated in our lab (Themis2 is closely related to Themis1, but is expressed in B cells and myeloid cells instead of T cells). Our results identified an important role for Themis2 in facilitating B cell activation by low-avidity, but not high-avidity, B cell receptor (BCR)–antigen interactions.
Figure 3. Themis is highly conserved in vertebrates.
Themis contains two novel CABIT domains, each with a conserved cysteine (red) and conserved flanking residues (yellow), a nuclear localization signal (NLS), and a proline-rich region (PRR).
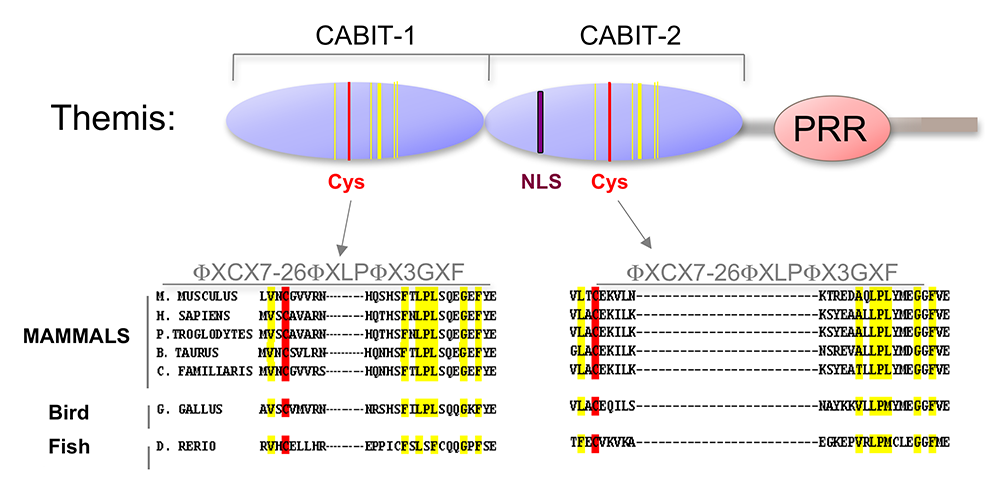
Figure 3. Themis is highly conserved in vertebrates.
Themis contains two novel CABIT domains, each with a conserved cysteine (red) and conserved flanking residues (yellow), a nuclear localization signal (NLS), and a proline-rich region (PRR).
More recently, we focused on determining the molecular function of Themis1. Themis1 (in addition to Themis2 and a large family of related metazoan proteins) contains a novel globular domain of previously unknown function called the CABIT (cysteine-containing, all beta in Themis) module (Figure 3). Using in vitro biochemical and protein-assay techniques, we determined that the Themis1 CABIT modules bind to the catalytic domain of SHP-1, a key hematopoietic protein tyrosine phosphatase. In the presence of reactive oxygen species (ROS), which are generated in activated T cells, Themis1, via its CABIT module, promoted oxidation of the redox-labile SHP-1 catalytic cysteine and therefore inactivated SHP-1. The CABIT modules from all five mammalian CABIT–containing proteins also inhibited SHP-1 (as well as several other protein tyrosine phosphatases), indicating that this activity was common to all CABIT modules. Given that SHP-1 is an inhibitory phosphatase that functions to dampen TCR signaling by de-phosphorylating several effector targets, including protein tyrosine kinases, the finding established an activating function for Themis1 in cell signaling through its ability to bind to and inhibit SHP-1. Interestingly, Themis1 is highly expressed in developing thymocytes at the stage at which they undergo positive selection. It had been known for years that thymocytes are more sensitive to TCR stimulation than are mature T cells, but the reason for this sensitivity was unknown. The function of Themis1, together with its high expression in thymocytes, provides an explanation for the increased sensitivity of thymocytes to TCR signaling (Figure 4). By showing that deletion of the gene encoding SHP-1 rescues T cell development in Themis1–/– mice, we confirmed that the primary role of Themis1 is to inhibit SHP-1. In addition to identifying the function of CABIT modules, our results provide insight into the role of other metazoan CABIT–containing proteins (which number in the hundreds). Our ongoing studies are focused on characterizing the intestine-specific CABIT protein Themis3, which is expressed in mice but not in humans, and on exploring a potential role of Themis proteins in human disease.
Figure 4. Regulated expression of Themis during T cell development facilitates thymocyte-positive selection.
Thymocytes are more sensitive to TCR signaling than mature T cells, exemplified by the fact that low-affinity self-ligands can activate thymocytes but are incapable of activating mature T cells. The relatively high expression of Themis in thymocytes compared with mature T cells contributes to establishing the sensitivity of thymocytes to TCR signaling. In thymocytes, in which Themis is abundant, Themis binds to and inactivates the inhibitory tyrosine phosphatase SHP-1 by promoting oxidation of the SHP-1 catalytic cysteine by reactive oxygen species (ROS, including H2O2), resulting in stronger TCR signaling to weak self-ligands. In mature T cells, down-regulation of Themis results in a higher concentration of active (reduced, i.e., catalytically active) SHP-1, which can inhibit TCR signals to low affinity self-ligands, preventing activation by self-ligands, which can result in autoimmunity.
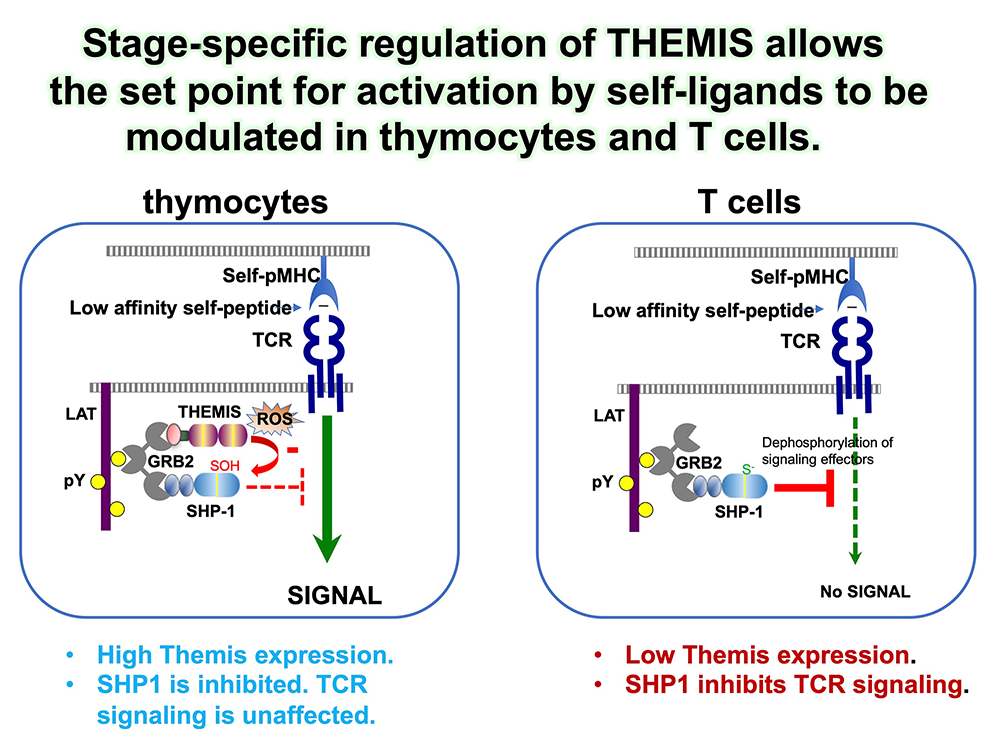
Figure 4. Regulated expression of Themis during T cell development facilitates thymocyte-positive selection.
Thymocytes are more sensitive to TCR signaling than mature T cells, exemplified by the fact that low-affinity self-ligands can activate thymocytes but are incapable of activating mature T cells. The relatively high expression of Themis in thymocytes compared with mature T cells contributes to establishing the sensitivity of thymocytes to TCR signaling. In thymocytes, in which Themis is abundant, Themis binds to and inactivates the inhibitory tyrosine phosphatase SHP-1 by promoting oxidation of the SHP-1 catalytic cysteine by reactive oxygen species (ROS, including H2O2), resulting in stronger TCR signaling to weak self-ligands. In mature T cells, down-regulation of Themis results in a higher concentration of active (reduced, i.e., catalytically active) SHP-1, which can inhibit TCR signals to low affinity self-ligands, preventing activation by self-ligands, which can result in autoimmunity.
Role of the F-box protein Fbxl12 in thymocyte development
An important element of the T cell maturation process is the precise regulation of cell proliferation. Rather than being a shared property among all or most developing thymocytes, proliferation is strictly limited to two stages during early T cell development. The initial proliferative phase is driven by thymus-expressed cytokines; the second coincides with ‘beta selection’ (i.e., is initiated in cells that have productively rearranged the TCRbeta chain gene and which then express a signaling complex called the pre–TCR). The proliferative burst that accompanies beta selection is estimated to result in a 100–200 fold expansion and is essential for further differentiation and for maximizing TCR diversity. Previous work showed that beta selection–associated proliferation requires concurrent signals by the pre–TCR and Notch receptors, but how these signals induce cell-cycle progression and why they need to be coordinated had remained unclear. Initiation of proliferation in beta-selected thymocytes requires the ubiquitin-mediated degradation of the cyclin-dependent kinase inhibitor Cdkn1b, which acts to prevent cell-cycle progression. In a recent study, we examined the molecular control of beta selection–associated proliferation. We confirmed prior findings that Cdkn1b degradation is induced by an SCF E3 ubiquitin ligase, which contains the ligand-recognition subunit Fbxl1. Deletion of Fbxl1 partially blocked beta selection–associated proliferation, a defect that was rescued by co-deletion of Cdkn1b. We identified a new F-box protein (the F-box protein domain mediates protein-protein interactions and ubiquitination), Fbxl12, that is highly expressed in immature thymocytes at the beta selection stage. We found that Fbxl12 also functions as an SCF E3 ligase subunit, which, like Fbxl1, directs Cdkn1b degradation. The phenotype of Fbxl12–deficient mice generated in our lab was strikingly similar to that of Fbxl1–deficient mice, and deletion of both Fbxl1 and Fbxl12 resulted in a more severe block in beta selection–associated proliferation (worse than single deletion of either Fbxl1 or Fbxl12), indicating that Fbxl1 and Fbxl12 act in concert to regulate thymocyte proliferation. Interestingly, we found that Fbxl1 expression is induced by Notch signaling, whereas Fbxl12 expression is induced by pre–TCR signaling. Both Fbxl1 and Fbxl12 are required for thymocyte proliferation; thus, their selective regulation by Notch and the pre–TCR, respectively, provides an explanation for why concurrent Notch and pre–TCR signaling is necessary for cell-cycle progression and proliferation at the beta-selection checkpoint. The requirement for concurrent pre–TCR and Notch signals to initiate optimal proliferation also ensures that proliferation of immature thymocytes only occurs when both signals are received, acting as a critical checkpoint in T cell development.
Role of Ldb1 transcription complexes in hematopoiesis and in the genesis of T cell acute lymphoblastic leukemia (T-ALL)
LIM domain binding protein-1 (Ldb1) is a ubiquitously expressed nuclear adapter protein that contains a LIM–zinc finger protein–interaction motif and a dimerization domain. In hematopoietic cells, Ldb1 functions by interacting with and/or by recruiting specific partners (including the LIM–only protein Lmo2 and the transcription factors Lyl1 or Tal1, and Gata1 or Gata2) to form multi-molecular transcription complexes (Figure 5). Within the hematopoietic lineage, expression of Ldb1 is highest in progenitor cells, which include hematopoietic stem cells (HSCs), but continues in all stages of T cell development. We initially investigated the role of Ldb1 in hematopoiesis by following the fate of Ldb1–/– embryonic stem cells (ESCs) in mouse blastocyst chimeras and by conditional, stage-specific deletion of Ldb1 in HSCs. We found that Ldb1 is not required for ESC maintenance but is required for HSC maintenance. More recent data indicate that the loss of Ldb1–/– HSCs results from defects in maintenance and differentiation. We performed a genome-wide screen for Ldb1–binding sites using ChIP-Seq. Analysis of the ChIP-Seq data revealed that Ldb1 complexes bind at the promoter or at regulatory sequences near a large number of genes known to be required for HSC maintenance. Examination of the function of Ldb1 in cell lineages downstream of HSCs identified an essential function in the erythroid lineage but not in myeloid or lymphoid cells. Further, ChIP-Seq analysis of Ldb1 DNA–binding complexes demonstrated that Ldb1 complexes in HSCs contain the transcription factor Gata2, whereas Ldb1 complexes in erythroid progenitors contain Gata1 (which is strongly expressed in the erythroid lineage). The results indicate that multimeric Ldb1 transcription complexes are modular and have distinct functions in the hematopoietic system depending on the cell type and their subunit composition, with Gata2–containing complexes regulating expression of HSC–maintenance genes in HSCs and Gata1 complexes regulating expression of erythroid-specific genes in erythroid cells (Figure 5). Current studies aim to determine how Ldb1 complexes regulate gene expression and the role of Ldb1 dimerization in mediating long-range promoter-enhancer interactions in hematopoietic cells. In addition, we are investigating a potential role for Ldb1 in regulating self-renewal of early T cell progenitors in the thymus.
Figure 5. Model of Ldb1 function in the hematopoietic lineage
Ldb1 forms a multimeric DNA–binding complex in hematopoietic cells with the adapter Lmo2 and the transcription factors Scl and Gata1 or Gata2. In hematopoietic stem cells (HSCs), in which Gata2 is highly expressed, Ldb1-Lmo2-Scl-Gata2 complexes positively regulate expression of HSC–maintenance genes. Differentiation of HSCs to the myeloid or lymphoid lineage (LMPP) is triggered by downregulation of Ldb1 complexes, whereas commitment to the erythroid lineage (ery) is triggered by induction of Gata1 and downregulation of Gata2, resulting in the formation of an Ldb1-Lmo2-Scl-Gata1 complex, which positively regulates expression of many erythroid-specific genes.
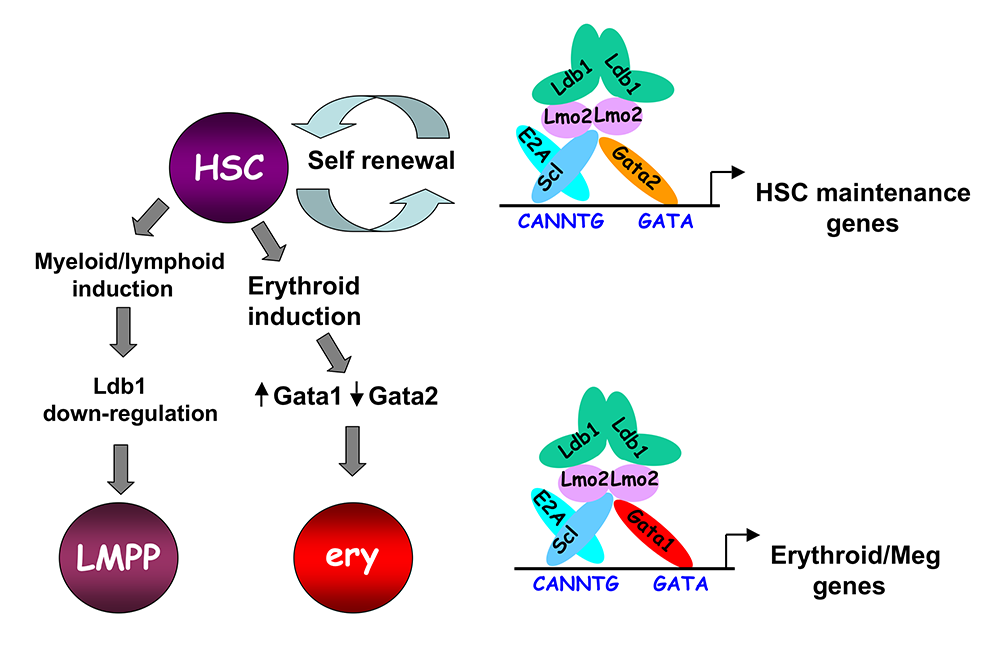
Figure 5. Model of Ldb1 function in the hematopoietic lineage
Ldb1 forms a multimeric DNA–binding complex in hematopoietic cells with the adapter Lmo2 and the transcription factors Scl and Gata1 or Gata2. In hematopoietic stem cells (HSCs), in which Gata2 is highly expressed, Ldb1-Lmo2-Scl-Gata2 complexes positively regulate expression of HSC–maintenance genes. Differentiation of HSCs to the myeloid or lymphoid lineage (LMPP) is triggered by downregulation of Ldb1 complexes, whereas commitment to the erythroid lineage (ery) is triggered by induction of Gata1 and downregulation of Gata2, resulting in the formation of an Ldb1-Lmo2-Scl-Gata1 complex, which positively regulates expression of many erythroid-specific genes.
Acute lymphoblastic leukemias (ALLs) are the most common type of cancer in children. T cell ALL (T-ALL) results from oncogenic transformation of immature intra-thymic T cell progenitors (thymocytes). Mouse models of T-ALL have been generated, one of the most informative being the Lmo2–transgenic (Lmo2-tg) mouse, which mis-expresses the nuclear adapter Lmo2 in thymocytes beyond the stage when it is normally down-regulated. The Lmo2-tg model closely mimics a prevalent type of human T-ALL, which is associated with chromosomal mutations that result in mis-expression of LMO2. It was recently reported that mis-expression of Lmo2 in mouse thymocytes beyond the early T progenitor (ETP) stage induces T-ALL at two distinct stages of development (the early ETP stage and a later ‘DN3’ stage). Notably, human T-ALLs can also occur at two similar stages of thymocyte maturation. The most immature forms of T-ALL that occur in Lmo2-tg mice and in humans express high levels of the transcription factor Hhex and are designated ETP T-ALLs, whereas later-stage T-ALLs do not over-express Hhex but express high levels of more mature markers of T cell development, including Notch1, Dtx1, Ptcra, and Hes1. Lmo2 functions as a critical anchoring subunit of the multimeric Ldb1–nucleated DNA–binding complexes described above. We found that normal ETP thymocyte progenitor cells express the same Ldb1 complex subunits that are present in HSCs, and it was shown by others that ETPs exhibit HSC characteristics, including self-renewal potential. Hhex is a target of Ldb1 complexes in HSCs and ETPs, a result that strongly suggests that Ldb1 complexes are responsible for the aberrant self-renewal in Lmo2-tg mice, which predisposes to oncogenesis. We hypothesized that Ldb1 complexes regulate self-renewal in ETPs as well as in HSCs. Lmo2 is normally down-regulated after the ETP stage when thymocytes undergo T lineage commitment, suggesting that extinguishing the expression of Lmo2 (and by extension, Ldb1 complexes) is important for extinguishing self-renewal potential and for T cell differentiation and that failure to do so predisposes to oncogenesis via ‘second-hit’ transforming events.
In RNA-Seq gene-expression experiments, we found that the RNA–expression signatures of Lmo2-tg immature thymocytes and HSCs are very similar, consistent with the notion that failure to downregulate Lmo2 ‘freezes’ cells in a stem-cell self-renewal state. To determine whether Ldb1 complexes are in fact required for abnormal thymocyte self-renewal, predisposing to ETP-ALL, and to explore the genes regulated by these complexes, we conditionally deleted Ldb1 in Lmo2-tg mice. We found that Ldb1 is required for Lmo2-tg–induced thymocyte self-renewal, expression of HSC renewal genes, and for T-ALL induction, indicating that Lmo2 mis-expression promotes T-ALL by functioning as a subunit of Ldb1 complexes (Figure 6). Currently, we are determining the subunit structure and binding sites of Ldb1 complexes expressed in Lmo2-tg thymocytes. We anticipate that our results will provide insights into the mechanisms controlling ETP-ALL oncogenesis in humans and may thus provide new therapeutic avenues for the treatment of this devastating, primarily pediatric disease.
Figure 6. Deletion of Ldb1 prevents Lmo2–induced generation of T cell leukemia.
Failure to down-regulate the Ldb1 complex subunit and proto-oncogene Lmo2 in mice and humans results in induction of a severe form of T cell leukemia (ETP-T-ALL), characterized by prolonged expression of hematopoietic stem-cell genes. Ldb1-Lmo2 multi-protein complexes are normally expressed in hematopoietic stem cells (HSCs) and function in the maintenance and self-renewal of HSCs (upper panel). Spontaneous mutations or retroviral insertions in humans or transgenic expression of Lmo2 in immature mouse thymocytes, promote abnormal self-renewal of thymocytes, rendering them sensitive to secondary oncogenic mutations, which result in induction of leukemia (T-ALL) (middle panel). Deletion of Ldb1 prevents induction of T-ALL in Lmo2 transgenic mice, identifying Ldb1 complexes as the cause of T-ALL in Lmo2–transgenic mice (lower panel).
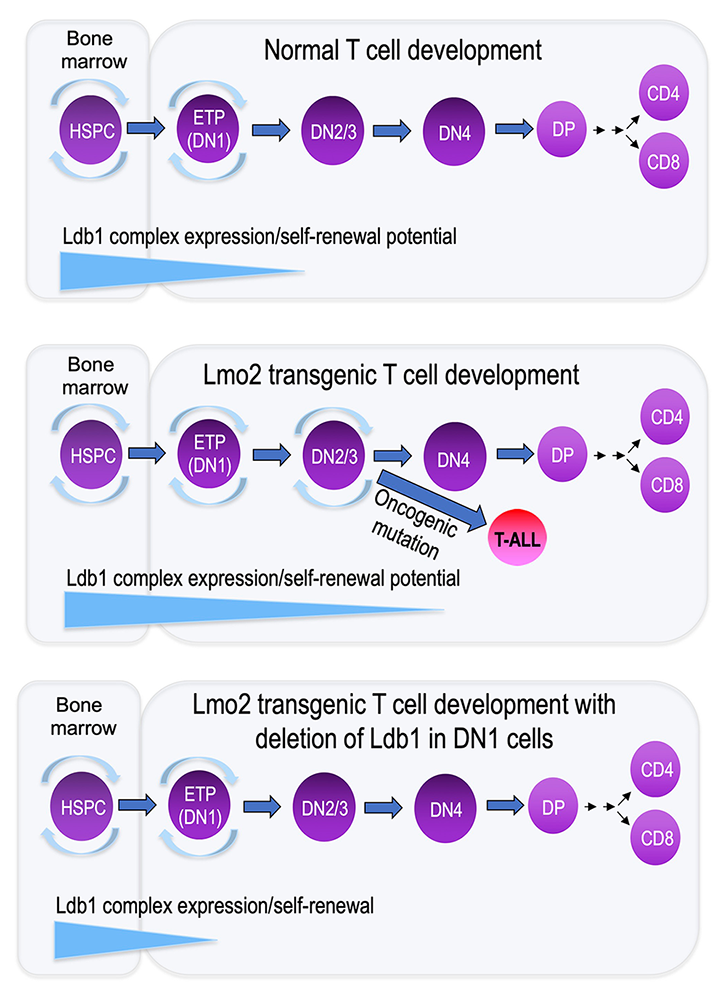
Figure 6. Deletion of Ldb1 prevents Lmo2–induced generation of T cell leukemia.
Failure to down-regulate the Ldb1 complex subunit and proto-oncogene Lmo2 in mice and humans results in induction of a severe form of T cell leukemia (ETP-T-ALL), characterized by prolonged expression of hematopoietic stem-cell genes. Ldb1-Lmo2 multi-protein complexes are normally expressed in hematopoietic stem cells (HSCs) and function in the maintenance and self-renewal of HSCs (upper panel). Spontaneous mutations or retroviral insertions in humans or transgenic expression of Lmo2 in immature mouse thymocytes, promote abnormal self-renewal of thymocytes, rendering them sensitive to secondary oncogenic mutations, which result in induction of leukemia (T-ALL) (middle panel). Deletion of Ldb1 prevents induction of T-ALL in Lmo2 transgenic mice, identifying Ldb1 complexes as the cause of T-ALL in Lmo2–transgenic mice (lower panel).
Additional Funding
- Fellows Award for Research Excellence (FARE) 2025 (TH)
Publications
- Innate immune-cell-derived BAFF promotes inflammatory response of ICOSL⁺ B cells via non-canonical NF-κB in diabetic autoimmunity. Mol Ther 2025 Dec 6:doi: 10.1016/j.ymthe.2025.12.003
- Early events in TCR signaling - the evolving role of ITAMs. Front Immunol 2025 16:1563049
- Engineering TCR-controlled fuzzy logic into CAR T cells enhances therapeutic specificity. Cell 2025 188:2372-2389
- Commensal-derived trehalose monocorynomycolate triggers γδ T cell-driven protective ocular barrier immunity. Immunity 2025 doi: 10.1101/2025.03.17.643820;preprint
- The common murine retroviral integration site activating Hhex marks a distal regulatory enhancer co-opted in human early T-cell precursor leukemia. J Biol Chem 2025 301(3):108233
Collaborators
- Grégoire Altan-Bonnet, PhD, Laboratory of Integrative Cancer Immunology, NCI, Bethesda, MD
- Remy Bosselut, MD, PhD, Laboratory of Immune Cell Biology, NCI, Bethesda, MD
- Richard J. Cornall, FRCP, FMedSci, Nuffield Department of Medicine, University of Oxford, Oxford, United Kingdom
- Utpal P. Davé, MD, Indiana University Melvin and Bren Simon Comprehensive Cancer Center, Indianapolis, IN
- James Gulley, MD, PhD, Center for Immuno-Oncology, Center for Cancer Research, NCI, Bethesda, MD
- Christian Hinrichs, MD, Rutgers Cancer Institute, New Brunswick, NJ
- Aravind Iyer, PhD, Protein and Genome Evolution Research Group, NLM/NCBI, Bethesda, MD
- Renaud Lesourne, PhD, INSERM, Toulouse, France
- Dorian McGavern, PhD, Viral Immunology and Intravital Imaging Section, NINDS, Bethesda, MD
- Paul Schumacker, PhD, Feinberg School of Medicine, Northwestern University, Chicago, IL
- Nevil Singh, PhD, University of Maryland School of Medicine, Baltimore, MD
- Naomi Taylor, MD, PhD, Pediatric Oncology Branch, Center for Cancer Research, NCI, Bethesda, MD
Contact
For more information, email lovep@mail.nih.gov or visit https://www.nichd.nih.gov/research/atNICHD/Investigators/love.